Inflammation and Oxidation in MPNs
Inflammation:
Abnormal expression and activity of a number of proinflammatory cytokines are implicated in MPNs (ref):
IL-1β, TNF-α, IL-6, IL-8, VEGF, PDGF, TGF-β and IFNs.
Chronic inflammation may be an important driving force for clonal evolution and disease progression in MPNs.
Tumor necrosis factor (TNFα), may underlie malignant clonal dominance in MPN based on results from mouse models. (ref)
In MF, bone marrow fibrosis itself is thought to be mediated heavily by the cytokine TGF-β, and possibly other cytokines produced as a result of hyperactivated JAK2 kinase in the malignant clone. (ref)
Treatment of MF with the TGF-β 1/3 Inhibitor AVID200 (MPN-RC 118) (ref)
Successful case report of controlling MF via TGF-β inhibiting supplements (ref)
Successful MPN treatments (INF-alpha, Ruxolitinib, Aspirin, Statins etc) also tend to be powerful anti-inflammatories.
Oxidation:
Reactive oxygen species (ROS) have a major role in carcinogenesis and disease progression in MPNs, where the malignant clone itself produces excess of ROS thereby creating a vicious self-perpetuating circle in which ROS activate proinflammatory pathways (NF-κB) which in turn create more ROS. (ref)
Targeting ROS via NF-κB inhibition may be a therapeutic option, which could possibly prevent genomic instability and ultimately myelofibrotic and leukemic transformation.
BET inhibitor, Pelabresib, being clinical trialed in MF is also a NF-KB inhibitor (ref).
Aspirin and ibuprofen are least potent, while resveratrol, curcumin, celecoxib, and tamoxifen are the most potent anti-inflammatory and antiproliferative agents of NF-kB. (ref)
Successful case report of treating MF with high dose Curcumin and COX2 inhibition (ref).
The gene NRF2 is significantly downregulated in all MPNs (ref). Activating NRF2 is a viable strategy to fight MPNs.
MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives (link)
Chronic inflammation may be an important driving force for clonal evolution and disease progression in MPNs. Abnormal expression and activity of a number of proinflammatory cytokines are associated with MPNs, in particular MF, in which immune dysregulation is pronounced as evidenced by dysregulation of several immune and inflammation genes. In addition, chronic inflammation has been suggested to contribute to the development of premature atherosclerosis and may drive the development of other cancers in MPNs, both nonhematologic and hematologic. The MPN population has a substantial inflammation-mediated comorbidity burden. Early intervention with interferon-alpha2, which as monotherapy has been shown to be able to induce minimal residual disease, in combination with potent anti-inflammatory agents such as JAK-inhibitors is foreseen as the most promising new treatment modality in the years to come.
Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms, 2021
Myeloid neoplasms, including acute myeloid leukemia (AML), myeloproliferative neoplasms (MPNs), and myelodysplastic syndromes (MDS), feature clonal dominance and remodeling of the bone marrow niche in a manner that promotes malignant over non-malignant hematopoiesis. This take-over of hematopoiesis by the malignant clone is hypothesized to include hyperactivation of inflammatory signaling and overproduction of inflammatory cytokines. In the Ph-negative MPNs, inflammatory cytokines are considered to be responsible for a highly deleterious pathophysiologic process: the phenotypic transformation of polycythemia vera (PV) or essential thrombocythemia (ET) to secondary myelofibrosis (MF), and the equivalent emergence of primary myelofibrosis (PMF). Bone marrow fibrosis itself is thought to be mediated heavily by the cytokine TGF-β, and possibly other cytokines produced as a result of hyperactivated JAK2 kinase in the malignant clone. MF also features extramedullary hematopoiesis and progression to bone marrow failure, both of which may be mediated in part by responses to cytokines. In MF, elevated levels of individual cytokines in plasma are adverse prognostic indicators: elevated IL-8/CXCL8, in particular, predicts risk of transformation of MF to secondary AML (sAML). Tumor necrosis factor (TNF, also known as TNFα), may underlie malignant clonal dominance, based on results from mouse models. Human PV and ET, as well as MF, harbor overproduction of multiple cytokines, above what is observed in normal aging, which can lead to cellular signaling abnormalities separate from those directly mediated by hyperactivated JAK2 or MPL kinases. Evidence that NFκB pathway signaling is frequently hyperactivated in a pan-hematopoietic pattern in MPNs, including in cells outside the malignant clone, emphasizes that MPNs are pan-hematopoietic diseases, which remodel the bone marrow milieu to favor persistence of the malignancy. Clinical evidence that JAK2 inhibition by ruxolitinib in MF neither reliably reduces malignant clonal burden nor eliminates cytokine elevations, suggests targeting cytokine mediated signaling as a therapeutic strategy, which is being pursued in new clinical trials. Greater knowledge of inflammatory pathophysiology in MPNs can therefore contribute to the development of more effective therapy.
Angela Fleischman, MD, PhD-Chronic Inflammation in MPNs (link)
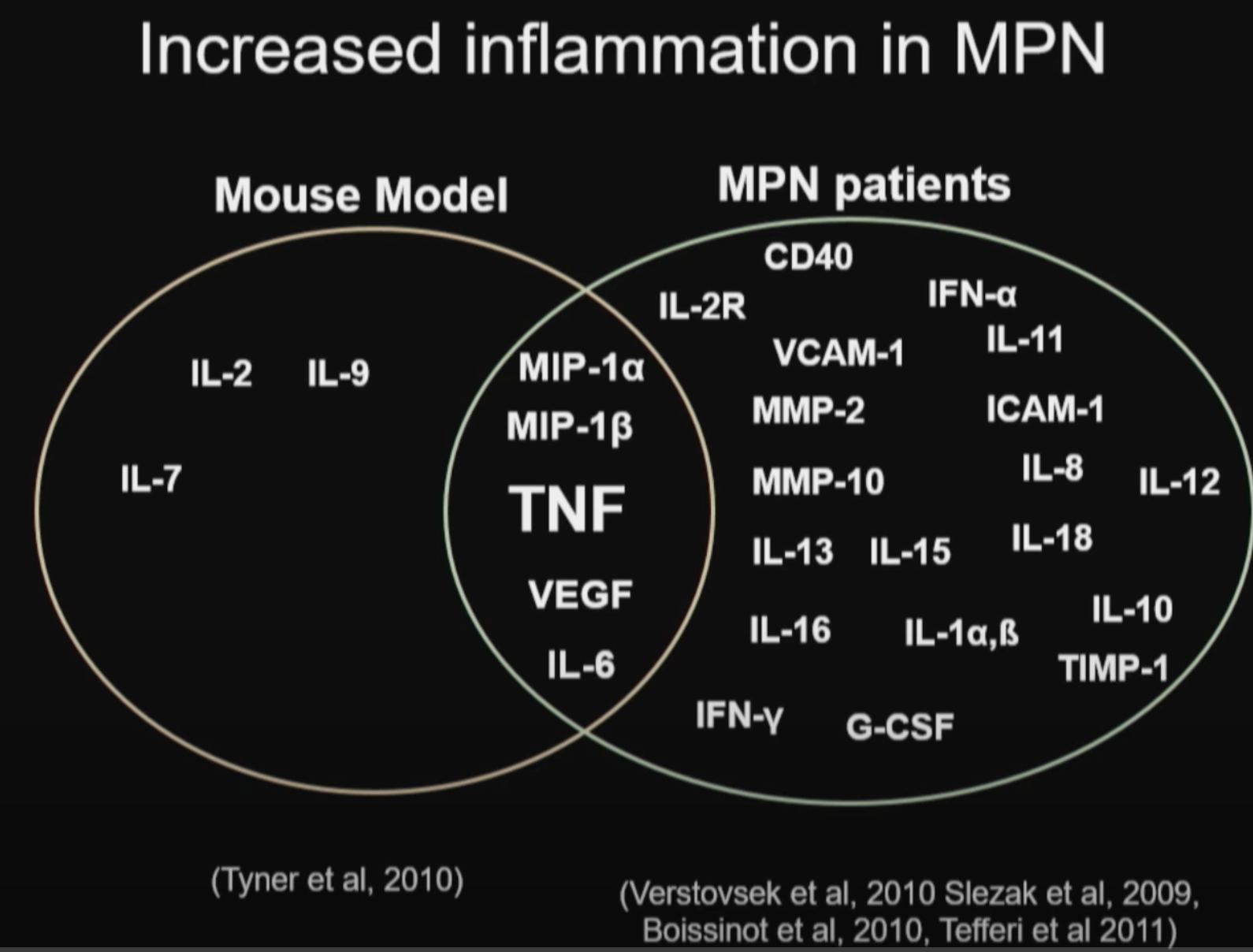
TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms (link)
Proinflammatory cytokines such as TNFα are elevated in patients with myeloproliferative
neoplasms (MPN), but their contribution to disease pathogenesis is unknown. Here we
reveal a central role for TNFα in promoting clonal dominance of JAK2V617F expressing …
Chronic inflammation: is it the driver or is it paving the road for malignant transformation? (link)
Chronic inflammation in well-defined mouse models such as Giα2 knock out mouse has
been shown to trigger formation and expansion of hypoxic niches and also leads to up
regulation of NFĸB, offering cells which have adapted their genetic machinery to hypoxia a …
The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms, 2015
Reactive oxygen species (ROS) have been implicated in a wide variety of disorders ranging between traumatic, infectious, inflammatory, and malignant diseases. ROS are involved in inflammation-induced oxidative damage to cellular components including regulatory proteins and DNA. Furthermore, ROS have a major role in carcinogenesis and disease progression in the myeloproliferative neoplasms (MPNs), where the malignant clone itself produces excess of ROS thereby creating a vicious self-perpetuating circle in which ROS activate proinflammatory pathways (NF-κB) which in turn create more ROS. Targeting ROS may be a therapeutic option, which could possibly prevent genomic instability and ultimately myelofibrotic and leukemic transformation. In regard to the potent efficacy of the ROS-scavenger N-acetyl-cysteine (NAC) in decreasing ROS levels, it is intriguing to consider if NAC treatment might benefit patients with MPN. The encouraging results from studies in cystic fibrosis, systemic lupus erythematosus, and chronic obstructive pulmonary disease warrant such studies. In addition, the antioxidative potential of the widely used agents, interferon-alpha2, statins, and JAK inhibitors, should be investigated as well. A combinatorial approach using old agents with anticancer properties together with novel JAK1/2 inhibitors may open a new era for patients with MPNs, the outlook not only being “minimal residual disease” and potential cure but also a marked improvement in inflammation-mediated comorbidities.

NF-κB, inflammation, immunity and cancer: coming of age (link)
Inflammation has been recognized as a hallmark of cancer and is known to play an essential role in the development and progression of most cancers, even those without obvious signs of inflammation and infection.
Nuclear factor-κB (NF-κB), a transcription factor that is essential for inflammatory responses, is one of the most important molecules linking chronic inflammation to cancer, and its activity is tightly regulated by several mechanisms.
Activation of NF-κB is primarily initiated by bacterial endotoxins such as lipopolysaccharide and pro-inflammatory cytokines such as tumour necrosis factor and IL-1. NF-κB activation occurs in cancer cells and in the tumour microenvironments of most solid cancers and haematopoietic malignancies.
NF-κB activation induces various target genes, such as pro-proliferative and anti-apoptotic genes, and NF-κB signalling crosstalk affects many signalling pathways, including those involving STAT3, AP1, interferon regulatory factors, NRF2, Notch, WNT–β-catenin and p53.
All known hallmarks of cancer involve NF-κB activation. In addition to enhancing cancer cell proliferation and survival, NF-κB and inflammation promote genetic and epigenetic alterations, cellular metabolic changes, the acquisition of cancer stem cell properties, epithelial-to-mesenchymal transition, invasion, angiogenesis, metastasis, therapy resistance and the suppression of antitumour immunity.
The prevalence of NF-κB activation in cancer-related inflammation makes it an attractive therapeutic target with the potential for minimal side effects.
A case of myelofibrosis controlled, 2017
“Fibrosis” begins with excessive deposition of extracellular matrix. That can evolve to fibrosis/scar tissue & considered cause of most organ failure
It is driven by excessive transforming growth factor beta and can involve genetic and epigenetic factors in the action pathway. The condition can be reversed, if TGF-beta production is decreased.
Initial thought was to treat myelofibrosis as fibrosis problem because of the failure from other therapy
Agents that lower TGF-β
Epigallocatechin gallate (EGCG) - Taurine
Metformin - Berberine
Curcumin - Quercetin
N-acetylcysteine - Lycopene
Silymarin - Glycyrrhiza
Cod liver oil - Ascorbic acid
Boswellic acids - Astaxanthin
Vitamin D - Hesperetin
Caffeine - Hesperidin
Phytolacca - Vitamin B6
References are available on PubMed
Treatment program
From physical signs of malabsorption and the low immune system, patient placed on GFD w/o test
Metformin-ER 500 mg bid – several anti-malignant actions, including in the hematopoietic area, can decrease cell proliferation encouraged by mTOR, and attenuate organ fibrosis
Berberine 500 mg bid - contributes to lowering the mTOR pathway implicated in myelofibrosis.
Quercetin 1000 mg bid – (Note: Transfusion frequency dramatically decreased after replacement by a highly absorbed form [caged molecule by Tesseract], approx. 100% delivery of 105 mg) Quercetin has many anti-cancer mechanisms. Known to lower TGFbeta and reduce fibrosis. Quercetin can inhibit the JAK/STAT cascade of inflammation and cell proliferation
Astaxanthin, 12 mg bid – quenches hydroxyl radical and possibly slows further DNA mutation
Vitamin K2 (15 mg daily as MK4) for sub-skin bleeding, due to platelets of 70
PubMed search - this seems the 1st report controlling myelofibrosis with refractory anemia (other than stem cell transplantation) and by mostly non-Rx agents.
See also link for the full paper.
Whole Blood Transcriptional Profiling Reveals Deregulation of Oxidative and Antioxidative Defence Genes in Myelofibrosis and Related Neoplasms. Potential Implications of Downregulation of Nrf2 for Genomic Instability and Disease Progression, 2014
The Philadelphia-negative chronic myeloproliferative neoplasms - essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF) (MPNs) - have recently been shown to be associated with chronic inflammation, oxidative stress and accumulation of reactive oxygen species (ROS). Using whole blood transcriptional profiling, we report that several oxidative stress and anti-oxidative stress genes are significantly deregulated in MPNs. Among the twenty most up- and downregulated genes, ATOX1, DEFB122, GPX8, PRDX2, PRDX6, PTGS1, and SEPP1 were progressively upregulated from ET over PV to PMF, whereas AKR1B1, CYBA, SIRT2, TTN, and UCP2 were progressively downregulated in ET, PV and PMF (all FDR <0.05). The gene Nrf2, encoding the transcription factor nuclear factor erythroid 2-related factor 2 (NFE2L2 or Nrf2) was significantly downregulated in all MPNs. Nrf2 has a key role in the regulation of the oxidative stress response and modulates both migration and retention of hematopoietic stem cells (HSCs) in their niche. The patogenetic importance of Nrf2 depletion in the context of expansion of the hematopoietic progenitor pool in MPNs is discussed with particular focus upon the implications of concomitant downregulation of Nrf2 and CXCR4 for stem cell mobilization.
NRF2's Role In Infections, Metabolism, & Longevity
https://mybiohack.com/blog/nrf2-cirs-sensitivities

Nrf2 affects the efficiency of mitochondrial fatty acid oxidation, 2014
Transcription factor Nrf2 (NF-E2 p45-related factor 2) regulates the cellular redox homoeostasis and cytoprotective responses, allowing adaptation and survival under conditions of stress. In the present paper, we report that the mitochondrial oxidation of long-chain (palmitic) and short-chain (hexanoic) fatty acids is depressed in the absence of Nrf2 and accelerated when Nrf2 is constitutively active. Addition of fatty acids stimulates respiration in heart and liver mitochondria isolated from wild-type mice. This effect is significantly weaker when Nrf2 is deleted, whereas it is stronger when Nrf2 activity is constitutively high. In the absence of glucose, addition of fatty acids differentially affects the production of ATP in mouse embryonic fibroblasts from wild-type, Nrf2-knockout and Keap1 (Kelch-like ECH-associated protein 1)-knockout mice. In acute tissue slices, the rate of regeneration of FADH2 is reduced when Nrf2 is absent. This metabolic role of Nrf2 on fatty acid oxidation has implications for chronic disease conditions including cancer, metabolic syndrome and neurodegeneration.

Surprising results of a supportive integrated therapy in myelofibrosis, 2014
Objectives: Myelofibrosis (MF) is characterized by shortened survival and a greatly compromised quality of life. Weight loss and cachexia seem to be the most important factors influencing survival in patients with MF. The aim of this study was to assess the efficacy of an integrated supportive therapy in improving cachexia and MF-related symptoms.
Methods: We reported on a case of a patient with MF who presented with weight loss and cachexia associated with severe anemia, fatigue, fever, and bone pain. The circulating levels of inflammatory, oxidative stress parameters, hepcidin, and erythropoietin were evaluated and were above normal ranges. The patient was treated with a multitargeted approach specifically developed for cachexia including oral l-carnitine, celecoxib, curcumin, lactoferrin, and subcutaneous recombinant human erythropoietin (EPO)-α.
Results: Surprisingly, after 1 y, cachexia features improved, all MF symptoms were in remission, and inflammatory and oxidative stress parameters, hepcidin, and EPO were reduced.
Conclusions: Because our protocol was targeted at inflammation and the metabolic state, its effectiveness may emphasize the role of inflammation in the pathogenesis of MF symptoms and demonstrates a need for the study of new integrated therapeutic strategies.

Curcumin Downregulates NF-kB and Related Genes in Patients with Multiple Myeloma: Results of a Phase I/II Study, 2007
High dose curcumin inhibiting Stat3 by 69% in blood mononuclear cells.
Long-term stabilisation of myeloma with curcumin, 2017
60 months stabilization of multiple myleloma with 8g/day curcumin (very high dose!)
Fighting Inflammation by Inhibiting NF-KB (link)
The Role of Nuclear Factor κB in the Interferon Response (link)
As illustrated in Fig. 2, IFNs activate not only the JAK/STAT signaling pathway but also the NF-κB signaling pathway. NF-κB regulates the expression of coding (mRNA) and noncoding (miRNA) genes as well as the cellular response to IFNs. Although STAT3, TYK2, PI3K, AKT, IKK, TRAF, and NIK have been identified in the IFN-activated NF-κB signaling pathway, the components of this signaling pathway have not been fully elucidated and could be cell context dependent. ChIP assays on the promoters regulated by NF-κB indicate that the outcome is highly dependent on the interplay between different STAT, NF-κB, and IRF proteins. The therapeutic effectiveness of IFN in cancer is often limited by IFN's inability to induce significant cell death because of the high constitutive NF-κB activity in tumor cells. Since there is a close relationship of NF-κB between inflammation, tumorigenesis, and the cellular response to IFN, we hypothesize that selectively targeting the NF-κB pathways may represent a novel strategy for enhancing IFN's therapeutic effectiveness and/or diminishing IFN's undesirable side effects. Inhibiting NF-κB activity in vitro sensitizes cells to IFN's apoptotic and antiviral activities. However, it will be important to determine whether inhibiting NF-κB activity in vivo will enhance IFN's antiviral and antitumor clinical activity.
Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation, 2004
Nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin have been shown to suppress transcription factor NF-kappaB, which controls the expression of genes such as cyclooxygenase (COX)-2 and cyclin D1, leading to inhibition of proliferation of tumor cells. There is no systematic study as to how these drugs differ in their ability to suppress NF-kappaB activation and NF-kappaB-regulated gene expression or cell proliferation. In the present study, we investigated the effect of almost a dozen different commonly used NSAIDs on tumor necrosis factor (TNF)-induced NF-kappaB activation and NF-kappaB-regulated gene products, and on cell proliferation. Dexamethasone, an anti-inflammatory steroid, was included for comparison with NSAIDs. As indicated by DNA binding, none of the drugs alone activated NF-kappaB. All compounds inhibited TNF-induced NF-kappaB activation, but with highly variable efficacy. The 50% inhibitory concentration required was 5.67, 3.49, 3.03, 1.25, 0.94, 0.60, 0.38, 0.084, 0.043, 0.027, 0.024, and 0.010 mM for aspirin, ibuprofen, sulindac, phenylbutazone, naproxen, indomethacin, diclofenac, resveratrol, curcumin, dexamethasone, celecoxib, and tamoxifen, respectively. All drugs inhibited IkappaBalpha kinase and suppressed IkappaBalpha degradation and NF-kappaB-regulated reporter gene expression. They also suppressed NF-kappaB-regulated COX-2 and cyclin D1 protein expression in a dose-dependent manner. All compounds inhibited the proliferation of tumor cells, with 50% inhibitory concentrations of 6.09, 1.12, 0.65, 0.49, 1.01, 0.19, 0.36, 0.012, 0.016, 0.047, 0.013, and 0.008 mM for aspirin, ibuprofen, sulindac, phenylbutazone, naproxen, indomethacin, diclofenac, resveratrol, curcumin, dexamethasone, celecoxib, and tamoxifen, respectively. Overall these results indicate that aspirin and ibuprofen are least potent, while resveratrol, curcumin, celecoxib, and tamoxifen are the most potent anti-inflammatory and antiproliferative agents of those we studied.
Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy, 2013
Intrinsic or acquired resistance to chemotherapeutic agents is a common phenomenon and a major challenge in the treatment of cancer patients. Chemoresistance is defined by a complex network of factors including multi-drug resistance proteins, reduced cellular uptake of the drug, enhanced DNA repair, intracellular drug inactivation, and evasion of apoptosis. Pre-clinical models have demonstrated that many chemotherapy drugs, such as platinum-based agents, antracyclines, and taxanes, promote the activation of the NF-κB pathway. NF-κB is a key transcription factor, playing a role in the development and progression of cancer and chemoresistance through the activation of a multitude of mediators including anti-apoptotic genes. Consequently, NF-κB has emerged as a promising anti-cancer target. Here, we describe the role of NF-κB in cancer and in the development of resistance, particularly cisplatin. Additionally, the potential benefits and disadvantages of targeting NF-κB signaling by pharmacological intervention will be addressed.
Other compounds that may be useful in increasing the sensitivity of cancers with constitutively active NF-κB to chemotherapeutic drugs are herbs with anti-inflammatory properties including the natural phenol curcumin and parthenolide which occurs in the plant feverfew (Patel et al., 2000). Curcumin down-regulates transcription factors important for cell growth and survival, through modulation of the NF-κB and PI3K/AKT pathways (Reuter et al., 2008). Curcumin alone or in combination with chemotherapy is effective both in vitro and in vivo in a number of cancer types, including melanoma (Odot et al., 2004). The side effects of curcumin appear to be limited, with high oral doses being tolerated with minimal toxicity, although abdominal complaints have been reported (Lao et al., 2006; Epelbaum et al., 2010). A phase I clinical trial of patients with multiple myeloma showed that both curcumin alone or in combination with bioperine, an alkaloid isolate from black pepper, decreased NF-κB levels in peripheral blood mononuclear cells (PBMCs) (NCT00113841). Similarly, in a phase II trial, curcumin decreased NF-κB levels in PBMCs and possessed biological activity in some patients with advanced pancreatic cancer (Dhillon et al., 2008).
Can NF-κB Be Considered a Valid Drug Target in Neoplastic Diseases? Our Point of View, 2020
Abstract: Multidrug resistance (MDR), of the innate and acquired types, is one of major problems in treating tumor diseases with a good chance of success. In this review, we examine the key role of nuclear factor-kappa B (NF-κB) to induce MDR in three tumor models characterized precisely by innate or acquired MDR, in particular triple negative breast cancer (TNBC), hepatocellular carcinoma (HCC), and acute myeloid leukemia (AML). We also present different pharmacological approaches that our group have employed to reduce the expression/activation of this transcriptional factor and thus to restore chemo-sensitivity. Finally, we examine the latest scientific evidence found by other groups, the most significant clinical trials regarding NF-κB, and new perspectives on the possibility to consider this transcriptional factor a valid drug target in neoplastic diseases.
NF-κB inhibitor with Temozolomide results in significant apoptosis in glioblastoma via the NF-κB(p65) and actin cytoskeleton regulatory pathways, 2020
Glioblastoma (GBM) is the most malignant brain tumor characterized by intrinsic or acquired resistance to chemotherapy. GBM tumors show nuclear factor-κB (NF-κB) activity that has been associated with tumor formation, growth, and increased resistance to therapy. We investigated the effect of NF-κB inhibitor BAY 11-7082 with Temozolomide (TMZ) on the signaling pathways in GBM pathogenesis. GBM cells and patient-derived GBM cells cultured in 3D microwells were co-treated with BAY 11-7082 and TMZ or BAY 11-7082 and TMZ alone, and combined experiments of cell proliferation, apoptosis, wound healing assay, as well as reverse-phase protein arrays, western blot and immunofluorescence staining were used to evaluate the effects of drugs on GBM cells. The results revealed that the co-treatment significantly altered cell proliferation by decreasing GBM viability, suppressed NF-κB pathway and enhanced apoptosis. Moreover, it was found that the co-treatment of BAY 11-7082 and TMZ significantly contributed to a decrease in the migration pattern of patient-derived GBM cells by modulating actin cytoskeleton pathway. These findings suggest that in addition to TMZ treatment, NF-κB can be used as a potential target to increase the treatment’s outcomes. The drug combination strategy, which is significantly improved by NF-κB inhibitor could be used to better understand the underlying mechanism of GBM pathways in vivo and as a potential therapeutic tool for GBM treatment.
Inhibition of NF-kB enhances chemotherapy induced apoptosis in ovarian cancer cells, 2005
Objectives: The NF-kB transcription factors can both promote cell survival and induce apoptosis depending on cell type and context. It is thought that inhibition of NF-kB may differentially affect tumors that retain wt-p53, as compared to tumors with mutant p53. We sought to determine if the NF-kB inhibitors curcumin, resveratrol, and pyrrolidine dithiocarbamate (PDTC) could enhance cisplatin induced apoptosis in ovarian cancer cells. Methods: Constitutive activation of endogenous NF-kB was determined in a panel of 8 ovarian cancer cell lines by immunoblotting for IKBα. NF-kB is present in the majority of cells as an inactive cytoplasmic precursor complexed with the inhibitory subunit IkBα. Upon activation of NF-kB, IkBα is degraded. Consequently, cells with constitutive activation of NF-kB have low IkBα expression levels. Cell lines with wt-p53 (A2780, OVCAR4) and mutant/null p53 (SKOV3, CaOV3) were treated with the NF-kB inhibitors (curcumin, resveratrol, and PDTC) alone, or in combination with cisplatin. Cell viability was determined by sulforhodamine. Apoptotic fraction was determined by staining with propidium iodide and analyzing subGo content by flow cytometry. NF-kB activation was determined by transfecting cells with a NF-kB-dependent reporter plasmid, and measuring luciferase activity. Results: The NF-kB inhibitors used in this study induced cell death in ovarian cancer cell lines with wt and mutant/null p53 expression, suggesting that constitutive NF-kB activity is required for survival. NF-kB inhibition following treatment with curcumin, resveratrol, and PDTC was confirmed using the NF-kB-dependent reporter assay. All NF-kB inhibitors enhanced cisplatin induced apoptosis. Conclusions: Most ovarian cancer patients die of chemoresistant recurrent disease. Conventional chemotherapeutic agents induce apoptosis, and resistance to chemotherapy can be promoted by constitutive activation of NF-kB. We have shown that inhibition of NF-kB enhances chemotherapy induced apoptosis in ovarian cancer cells regardless of p53 status. These results suggest that the use of NF-kB inhibitors in combination with conventional chemotherapy may have broad clinical benefits to ovarian cancer patients.
Effectiveness of Curcumin for Treating Cancer During Chemotherapy, 2018
Oncologists tend to avoid the use of supplements during chemotherapy. To understand the safety issue, a literature review was conducted to evaluate safety and possible benefits of taking curcumin during chemotherapy. Curcumin was chosen because of its wide use in the public and because of the availability of clinical trials utilizing curcumin during chemotherapy. Curcumin is considered a “pan-assay interference compound,” creating false leads in drug discovery assays. The pharmacodynamics of curcumin presents many challenges for a therapeutic agent, including poor bioavailability and rapid metabolism and excretion. The proposed molecular targets for curcumin include inhibition of nuclear factor kappa B and inhibition of cyclooxygenase-2. Clinical trials investigating the efficacy of curcumin treatment for cancer have been conducted in patients with colorectal cancer (CRC), pancreatic cancer, breast cancer, prostate cancer, multiple myeloma, lung cancer, and head and neck cancer. Outcomes revealed that while curcumin was not effective, it was well tolerated and safe. Recently, there have been several Phase I and Phase II trials combining curcumin with chemotherapeutic agents. Two trials investigated the effect of curcumin given concurrently with gemcitabine for advanced pancreatic cancer patients. One trial investigated the effect of curcumin given concurrently with docetaxel in advanced breast cancer patients. Another trial investigated the role of curcumin given concurrently with docetaxel and prednisone in castration-resistant prostate cancer patients. The results again indicate the safety of curcumin, but no added benefit of adding curcumin to the chemotherapy regimen. The literature review demonstrated that curcumin is well tolerated up to 6–8 g in research. While there are many promising in vivo and in vitro trials on the potential benefits of curcumin for treatment of cancer in conjunction with chemotherapy, to date there is no evidence that combining curcumin with chemotherapeutic agents is effective for treating cancer in humans. The lack of effect may be due to the target diseases that are treatment resistant such as advanced pancreatic cancer and advanced breast cancer. Also, curcumin has many difficult properties such as poor solubility and rapid metabolism. Currently, there are developments of analogues, liposomal products, and nanoparticles that may overcome the current challenges of curcumin. Future studies should utilize these evolving technologies.
Strategies to improve the antiglioma action of IFN: a role for NF-kB inhibition
Brain tumors are among the leading causes of cancer-related deaths in the United States, with glioblastoma multiforme (GBM) being one of the most aggressive and difficult subtypes to treat. In this proposal we plan to pursue a strategy to treat malignant glioma with the potent antitumor cytokine, interferon-beta (IFN2). Type I (1/2) interferons (IFNs) have long been recognized for their significant, pleiotropic anticancer activity. However, despite significant activity in preclinical models against a variety of tumor types, including gliomas, the antitumor efficacy of IFNs in clinical trials has been disappointing. A significant contributing factor includes the development of resistance to IFN-mediated cell death through downregulation of apoptotic pathways. We have established that nuclear factor kB (NFkB) promotes cell survival and suppresses the expression of a subset of IFN target genes that are likely effectors of IFN's antitumor activity. Unfortunately, not only is NFkB constitutively active in many cancers, including glioma, but it can also be activated by IFN itself. This finding suggests that the potent anticancer activity of IFN may be counterbalanced by NFkB activity. Based on these observations, we hypothesize that selective inhibition of NFkB will enhance the anticancer activity of IFN. We will perform a systematic and detailed evaluation of the role of NFkB in regulating the anticancer action of IFN2 in glioma cells. Based on the insights gained, we will select and test complementary agents that should provide synergistic antitumor activity with IFN. After confirming the synergy of these agents in vitro, we will test the effectiveness of combination therapy that includes IFN in relevant preclinical models of malignant glioma. The overriding goal of this project is to increase the antitumor activity of IFN through an improved understanding of IFN2's mechanism of action against glioma and the factors that work against it.
In specific aim 1 we will examine the role of NFkB in suppressing the anticancer activity of IFN2 in gliomas. To test this, we will determine in glioma cell lines 1) the contribution of the classical NF:B pathway to constitutive and IFN-induced NF:B activity;2) the contribution of the alternative NF:B pathway to constitutive and IFN-induced NF:B activity;and 3) the effects of pharmacological and genetic NF:B inhibitors on IFN activity. Based on these findings, we will test the effects of clinically available NFkB inhibitors on IFN's anticancer activity in relevant, preclinical rodent models of malignant glioma.
In specific aim 2 we will characterize the role of NFkB in regulating IFN target genes and the role of these target genes in the anticancer action of IFN2 in gliomas. To test this, we will: 1) characterize the induction of these genes in response to IFN;2) determine their importance in effecting the anticancer activity of IFN in vitro and in vivo and 3) translate these findings by testing the efficacy of appropriately selected combination therapy in relevant, preclinical models of malignant glioma.
Inhibition of NF-κB Signaling Reduces the Stemness Characteristics of Lung Cancer Stem Cells, 2018
Cancer stem cells (CSCs) are a subpopulation of cancer cells that play a pivotal role in tumor development, invasion, metastasis, and recurrence. We and others have reported significant involvement of the NF-κB pathway in regulating CSCs of non-small cell lung cancer (NSCLC). In this study, we evaluated the effects of NF-κB inhibition on self-renewal, stemness, migration, and expression of genes involved in the epithelial to mesenchymal transition (EMT) and apoptosis resistance in lung CSCs. Different concentrations of the NF-κB inhibitor BMS-345541 (0.4, 4.0, and 10.0 µM), an inhibitor the NF-κB upstream kinase IKKβ, were used to treat both lung CSCs (CD166+CD44+, CD166+EpCAM+) and non-CSC NSCLC cells (CD166−CD44−, CD166−EpCAM−) in A549 and H2170 cell lines. We assessed the impact of BMS-345541 on the ability to form tumorspheres (self-renewal assay), expression of stemness genes (SOX2, OCT4, NANOG, SCA-1, and KLF4), migration, and expression of EMT and apoptosis-related genes. Inhibition of NF-κB by BMS-345541 effectively reduced the stemness, self-renewal, and migration capacity of lung CSCs. Moreover, expression of genes involved in the EMT (SNAI1 and TWIST) and apoptosis resistance (BCL-2, BAX, and BIRC5) was significantly reduced following the treatments, suggesting that NF-κB inhibition is sufficient to prevent the EMT and induce apoptosis in lung CSCs. Our findings suggest that NF-κB inhibition could reduce the capability of CSCs to maintain their population within the tumor mass, potentially decelerating cancer progression, relapse, and chemotherapy resistance.
Curcumin inhibits interferon-alpha induced NF-kappaB and COX-2 in human A549 non-small cell lung cancer cells, 2005
The A549 cells, non-small cell lung cancer cell line from human, were resistant to interferon (IFN)-alpha treatment. The IFN-alpha-treated A549 cells showed increase in protein expression levels of NF-kappaB and COX-2. IFN-alpha induced NF-kappaB binding activity within 30 min and this increased binding activity was markedly suppressed with inclusion of curcumin. Curcumin also inhibited IFN-alpha-induced COX-2 expression in A549 cells. Within 10 min, IFN-alpha rapidly induced the binding activity of a gamma-(32)P-labeled consensus GAS oligonucleotide probe, which was profoundly reversed by curcumin. Taken together, IFN-alpha-induced activations of NF-kappaB and COX-2 were inhibited by the addition of curcumin in A549 cells.
EBV induces persistent NF-κB activation and contributes to survival of EBV-positive neoplastic T- or NK-cells
Epstein–Barr virus (EBV) has been detected in several T- and NK-cell neoplasms such as extranodal NK/T-cell lymphoma nasal type, aggressive NK-cell leukemia, EBV-positive peripheral T-cell lymphoma, systemic EBV-positive T-cell lymphoma of childhood, and chronic active EBV infection (CAEBV). However, how this virus contributes to lymphomagenesis in T or NK cells remains largely unknown. Here, we examined NF-κB activation in EBV-positive T or NK cell lines, SNT8, SNT15, SNT16, SNK6, and primary EBV-positive and clonally proliferating T/NK cells obtained from the peripheral blood of patients with CAEBV. Western blotting, electrophoretic mobility shift assays, and immunofluorescent staining revealed persistent NF-κB activation in EBV-infected cell lines and primary cells from patients. Furthermore, we investigated the role of EBV in infected T cells. We performed an in vitro infection assay using MOLT4 cells infected with EBV. The infection directly induced NF-κB activation, promoted survival, and inhibited etoposide-induced apoptosis in MOLT4 cells. The luciferase assay suggested that LMP1 mediated NF-κB activation in MOLT4 cells. IMD-0354, a specific inhibitor of NF-κB that suppresses NF-κB activation in cell lines, inhibited cell survival and induced apoptosis. These results indicate that EBV induces NF-κB-mediated survival signals in T and NK cells, and therefore, may contribute to the lymphomagenesis of these cells.
NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells
Epstein-Barr virus (EBV) transforms B lymphocytes into lymphoblastoid cell lines usurping the Notch and tumor necrosis factor receptor pathways to effect transcription including NF-kappaB activation. To determine whether NF-kappaB activity is essential in the growth and survival of EBV-transformed lymphoblastoid cell lines, a nondegradable IkappaBalpha mutant was expressed under tetracycline regulation. Despite continued Bcl-2 and Bcl-x/L expression, NF-kappaB inhibition induced apoptosis as evidenced by poly(ADP-ribose) polymerase cleavage, nuclear condensation and fragmentation, and hypodiploid DNA content. Both caspase 3 and 8 activation and loss of mitochondrial membrane potential were observed in apoptotic cells. However, caspase inhibition failed to block apoptosis. These experiments indicate that NF-kappaB inhibitors may be useful in the therapy of EBV-induced cellular proliferation.
Hostile takeovers: viral appropriation of the NF-kB pathway
Transcriptional regulators of the NF-kB/IkB family promote the expression of well over 100 target genes, the majority of which participate in the host immune response (1). These proteins include a multitude of cytokines and chemokines, receptors required for immune recognition, proteins involved in antigen presentation, and adhesion receptors involved in transmigration across blood vessels walls. Because of this extensive role in immune action, NF-kB has been termed the central mediator of the immune response. Gene knockout and other studies establish roles for NF-kB in the ontogeny of the immune system but also demonstrate that NF-kB participates at multiple steps during oncogenesis (2) and the regulation of programmed cell death (3).
For several reasons, the NF-kB pathway provides an attractive target to viral pathogens. Activation of NF-kB is a rapid, immediate early (IE) event that occurs within minutes after exposure to a relevant inducer, does not require de novo protein synthesis, and results in a strong transcriptional stimulation of several early viral as well as cellular genes. In this review, we will describe strategies that viruses have evolved to modulate the NF-kB pathway, to enhance viral replication, host cell survival, and evasion of immune responses. Activation of NF-kB constitutes an obvious target because many of its target genes — growth factors, cytokines and their receptors, and proto-oncogenes — profoundly influence the host cell cycle. In addition, some viruses exploit the antiapoptotic properties of NF-kB to evade the host defense mechanisms that limit replication by killing infected cells, or conversely to trigger apoptosis as a mechanism to increase virus spread.
Perhaps not surprisingly, the persistent activation of the NF-kB pathway maintained by certain viruses contributes to oncogenic transformation (2). In addition to the classic studies with the avian REV-T retrovirus which contains the v-Rel oncoprotein and induces a rapid and fatal B-cell lymphoma in young birds (4), several lines of evidence demonstrate that NF-kB family members contribute to human oncogenesis. Localization of NF-kB–encoding genes at sites of chromosomal translocations and genomic rearrangements in cancer, high levels of NF-kB activity in many breast cancer cells, and constitutive nuclear NF-kB complexes in Hodgkin’s lymphoma cells all support this view (2). Furthermore, as discussed below, viral oncogene products, including human T-cell leukemia virus type 1 (HTLV-1) Tax protein and Epstein-Barr virus latent infection membrane protein 1 (EBV LMP1), each act by unique mechanisms to disrupt NF-kB regulation and initiate viral transformation.
WHICH INFLAMMATION TESTS DO I NEED? (2022)


Comments