PAI-1
Nattokinase <-> PAI-1
How Nattokinase does its magic (2017): Inhibition of PAI-1.
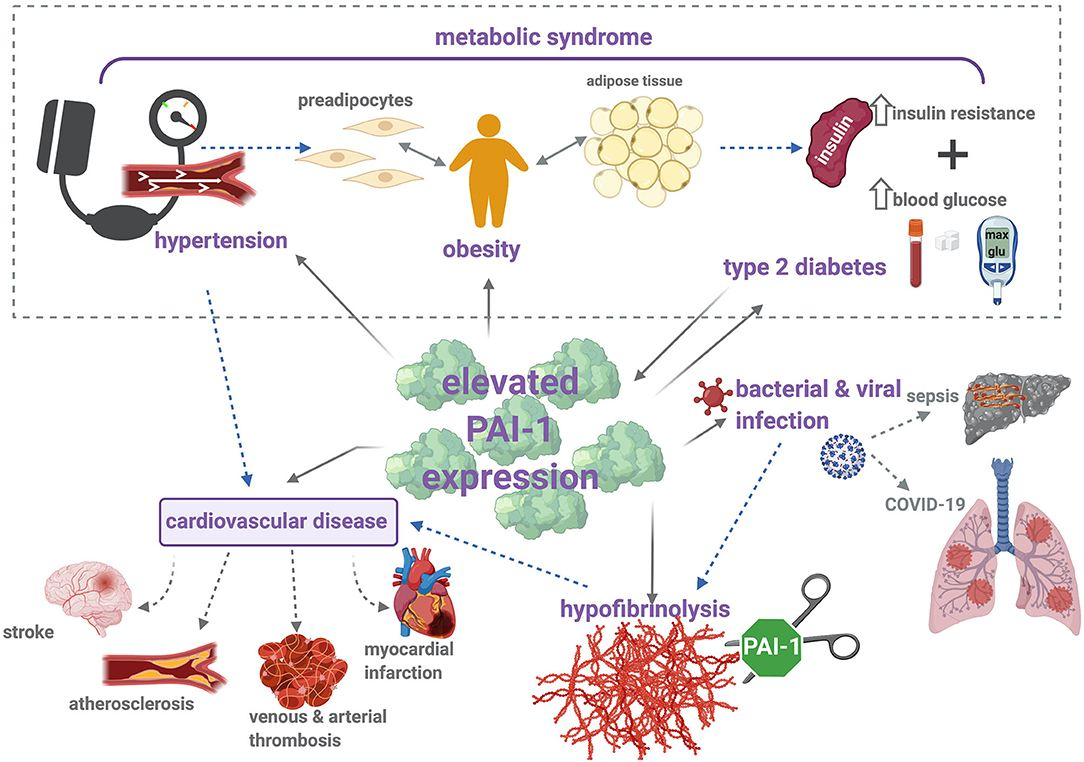
Elevated PAI-1 predicts adverse outcomes in many conditions - MI, stroke, cancer, diabetes, pregnancy complications. It is considered a promising biomarker linking thrombosis, inflammation, metabolic disorders, and aging (2010).
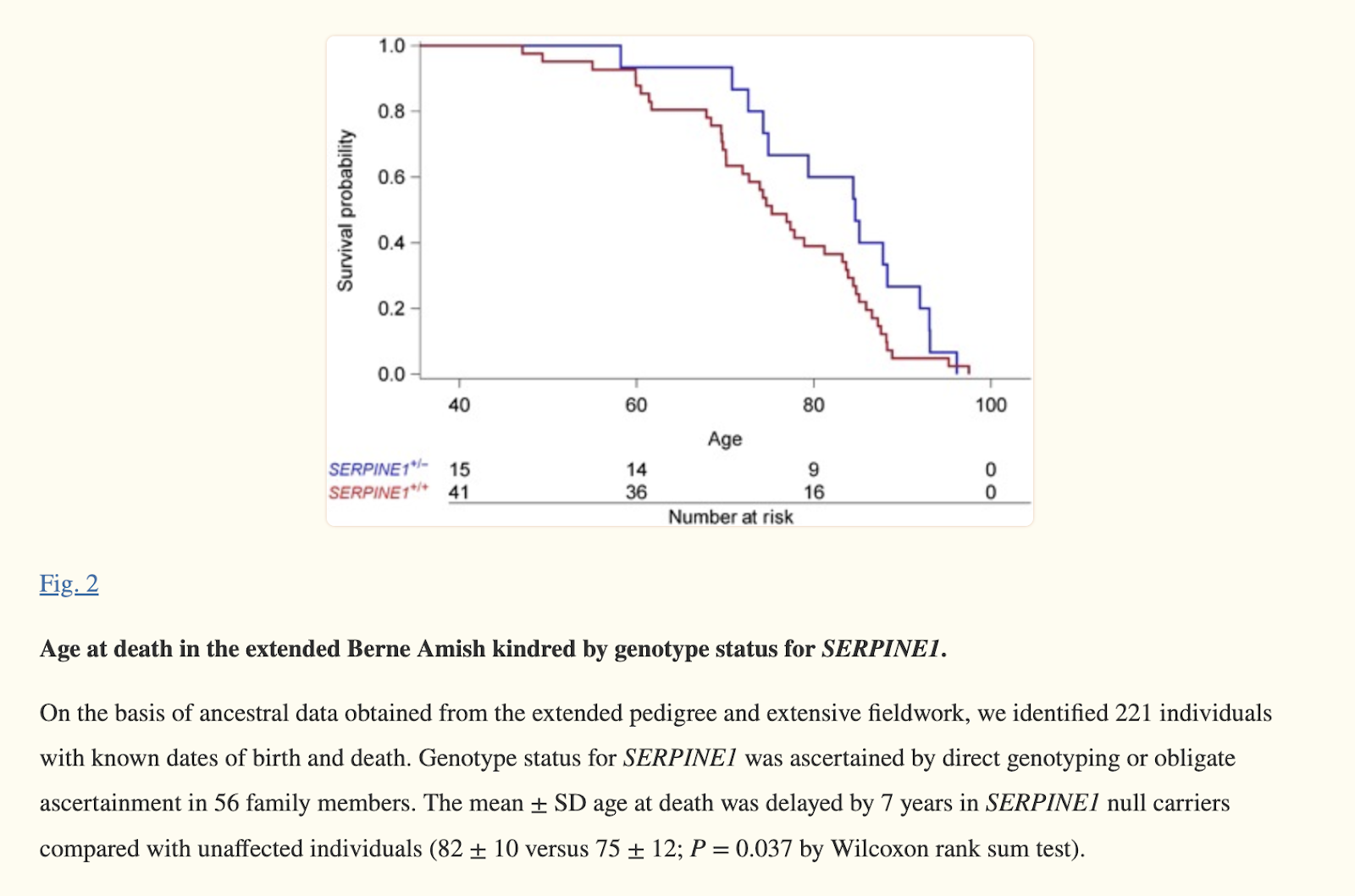
People who genetically doesn’t make too much PAI-1 live on average 7 yrs longer (2017).
Metabolic Syndrome is associated with an overproduction of PAI-1 (2006).
Inhibiting PAI-1 significantly helps with fatty liver and dyslipidemia. Activates FGF21 5-fold — a longevity molecule. And reduces PCKS9 (2021).
Giving natto bacteria to worms makes the worms live longer (2023).
Several drugs are in the clinical pipeline to inhibit PAI-1, but high dose nattokinase might be the safest option today.
Experimental drug (TM5614) reverses high cholesterol, obesity-related nonalcoholic fatty liver disease in animal study (2019)
Scientists at Northwestern University Feinberg School of Medicine discovered a novel molecular pathway involving the enzyme plasminogen activator inhibitor 1 (PAI-1) that plays a direct role in high cholesterol and nonalcoholic fatty liver disease (Levine et al., 2019).
They tested an investigational oral drug called TM5614 that inhibits PAI-1 in mice fed a high-fat, high-sugar diet to induce obesity.
After 10 days of treatment with TM5614, the obese mice showed significantly improved fasting blood sugar, insulin, and LDL cholesterol compared to untreated obese mice
The treated mice also exhibited a "remarkable" reduction in fatty liver disease
Further analysis revealed that PAI-1 prevents the inactivation of PCSK9, a protein that regulates cholesterol. TM5614 was able to rescue the ability to inactivate PCSK9.
The researchers suggest TM5614 could potentially provide a less expensive and easier oral alternative to PCSK9 inhibitor injections currently used to treat high cholesterol when statins are not enough.
Role of PAI-1 in hepatic steatosis and dyslipidemia (2021)
Plasminogen activator inhibitor 1 (PAI-1) is elevated in obesity, diabetes, and metabolic syndrome. Prior studies show PAI-1 contributes to obesity, insulin resistance, and fatty liver disease. This study aimed to investigate the mechanistic role of PAI-1 in lipid metabolism and dyslipidemia.
Pharmacological inhibition of PAI-1 with TM5614 in mice fed a chow diet led to reduced plasma PCSK9 levels (-40%) and total cholesterol (-25%). RNA sequencing of the liver showed Pcsk9 was the most downregulated gene (-86%) and Fgf21 the most upregulated (+568%) with PAI-1 inhibition.
PAI-1 heterozygous deficient (Pai-1+/-) mice also had alterations in hepatic lipid metabolism gene expression compared to wild-type, with Pcsk9 again among the most downregulated. Pai-1+/- mice had trends for reduced plasma PCSK9 (-16%, p=0.28) and total cholesterol (-16%, p=0.059).
In dyslipidemic mice fed a high-fat high-sugar (HFHS) diet, PAI-1 inhibition with TM5614 for 10 weeks reduced active plasma PAI-1 by 56% and total cholesterol by 37%, driven by reductions in LDL-C (-48%) and HDL-C (-33%). Hepatic Pcsk9 mRNA decreased by 81%, along with decreases in Srebp1a, Srebp1c, Srebp2, and HmgCoAr. Plasma PCSK9 decreased by 55%.
Pai-1+/- mice on a HFHS diet had 50% lower plasma PAI-1 and 20% lower total cholesterol compared to wild-type, driven by decreases in LDL-C and HDL-C. Hepatic Pcsk9 mRNA decreased by 50% and plasma PCSK9 by 61% with Pai-1 heterozygosity.
In humans, Amish individuals with a PAI-1 loss-of-function mutation had significantly lower plasma PCSK9 levels compared to unaffected controls (24.8 vs 41.2 ng/mL, p=0.02). PAI-1 and PCSK9 levels were positively correlated (r=0.557, p<0.001).
In a separate cohort of patients with heart failure, PAI-1 and PCSK9 levels were again positively correlated (p<0.0001).
Hypercholesterolemic patients treated with PCSK9 inhibitor antibodies had increased plasma PCSK9 but decreased plasma PAI-1 levels, suggesting a reciprocal regulatory relationship.
In conclusion, PAI-1 has a direct role in regulating hepatic lipid metabolism and cholesterol levels through effects on PCSK9 and other genes. Reducing PAI-1 activity lowers PCSK9 transcription and plasma levels in mice and humans. These findings identify PAI-1 inhibition as a potential therapeutic approach for dyslipidemia and prevention of cardiovascular complications of metabolic syndrome.
Role of PAI-1 in hepatic steatosis and dyslipidemia (2021)
Levine et al. (2021) demonstrated that plasminogen activator inhibitor-1 (PAI-1) is a critical regulator of hepatic lipid metabolism and a mechanistic contributor to dyslipidemia. The study used pharmacological inhibition and genetic deficiency of PAI-1 in mice to show that reducing PAI-1 activity leads to significant changes in the expression of numerous genes involved in lipid metabolism in the liver.
Specifically, RNA sequencing analysis found that short-term administration of the PAI-1 inhibitor TM5614 to mice on a standard chow diet resulted in Pcsk9 being the most downregulated hepatic gene transcript (86% decrease, p=4.34 x 10-32) while Fgf21 was the most upregulated gene (568% increase, p=5.66 x 10-28). Gene ontology analysis revealed robust alterations in the expression of 160 genes involved in lipid metabolism and 156 genes involved in fatty acid metabolism. The reduction in Pcsk9 expression was associated with a 40% decrease in plasma PCSK9 levels (p<0.05) and a 25% reduction in total plasma cholesterol levels (p<0.01), driven by decreased HDL cholesterol. Similar transcriptional changes in lipid metabolism genes, including an 82% downregulation of Pcsk9, were observed in Pai-1 heterozygous mice compared to wildtype controls.
To investigate further, the PAI-1 inhibitor TM5614 was administered to mice with diet-induced obesity and hyperlipidemia. This led to an 81% reduction in hepatic Pcsk9 mRNA expression (p<0.001), along with decreased expression of other regulators of cholesterol synthesis like Srebp1a, Srebp1c, and HMG-CoA reductase. There was a 55% decrease in plasma PCSK9 levels (p<0.01) and reductions in total cholesterol (37%, p<0.05), LDL cholesterol (48%, p=0.06), and HDL cholesterol (33%, p<0.05). Pai-1 heterozygous mice fed a high fat/high sugar diet also showed 50% lower hepatic Pcsk9 expression (p<0.05) and 20% lower total plasma cholesterol (p<0.05) compared to wildtype controls.
Analysis of plasma samples from humans with a PAI-1 loss-of-function mutation showed they had significantly lower plasma PCSK9 levels compared to unaffected individuals (p=0.02), and PCSK9 levels positively correlated with PAI-1 levels (r=0.557, p<0.001). A similar correlation was found between PAI-1 and PCSK9 levels in plasma samples from patients without PAI-1 mutations. Additionally, treatment of hypercholesterolemic patients with PCSK9 inhibitor antibodies led to increased PCSK9 levels but decreased PAI-1 levels in plasma.
In vitro experiments treating HepG2 cells with the PAI-1 inhibitor TM5614 resulted in decreased intracellular and extracellular PCSK9 protein levels, along with increased LDL receptor protein levels. This supports the conclusion that PAI-1 regulation of PCSK9 impacts cholesterol clearance mediated by LDL receptors.
In summary, using pharmacological inhibition and genetic models in mice, along with human plasma samples, Levine et al. demonstrated that PAI-1 activity directly regulates the expression of key lipid metabolism genes like Pcsk9. Reducing PAI-1 led to lower plasma cholesterol levels, likely due to increased LDL receptor-mediated clearance resulting from decreased PCSK9. The study revealed a novel role for PAI-1 as a regulator of hepatic lipid metabolism and contributor to dyslipidemia, providing insight into the pathogenesis of metabolic disease. Given that PAI-1 levels are elevated in obesity and metabolic syndrome, targeting PAI-1 may be a therapeutic strategy to lower cholesterol and potentially reduce cardiovascular complications.
Impaired fibrinolysis without hypercoagulability characterises patients with non-alcoholic fatty liver disease (2022)
Introduction: Cardiovascular disease is the major cause of mortality in non-alcoholic fatty liver disease (NAFLD), a disease affecting one quarter of the world's population. Coagulation imbalance may be a contributing factor but is yet to be convincingly disclosed.
Aim: To perform an extensive mapping of the hemostatic system; primary and secondary hemostasis and the fibrinolytic system in non-diabetic NAFLD patients.
Materials and methods: Twenty-five non-diabetic, biopsy-proven NAFLD patients [12 simple steatosis; 13 non-alcoholic steatohepatitis (NASH)] investigated by a comprehensive panel of coagulation and fibrinolysis tests in a cross-sectional study. Fifty age- and sex-matched healthy persons served as controls for each of the dynamic analyses: platelet aggregation, thrombin generation, fibrin formation and lysis. Body composition, insulin resistance makers, and liver fat assessed by proton density magnetic resonance imaging were measured in the patients.
Results: Fibrinolytic function was impaired in simple steatosis [median 50% clot lysis time 1123 (min-max, 618-1967) s] and NASH [1448 (521-2618) s] compared to healthy controls [403 (184-1179) s] (p < 0.0001). Plasminogen activator inhibitor-1 (PAI-1) increased stepwise above reference interval from simple steatosis [54 (29-80) ng/ml] to NASH patients [109 (65-153) ng/ml; p = 0.03]. Impaired fibrinolysis correlated with hepatic fat fraction and insulin resistance; PAI-1 correlated with obesity and insulin resistance (ρ ≥ 0.42; p ≤ 0.04). Platelet aggregation, coagulation factors, natural anticoagulants, and thrombin generation were comparable to healthy controls and established reference intervals.
Conclusions: NAFLD patients had impaired fibrinolysis without significant prothrombotic changes in coagulation. The impact of this abnormality on the increased cardiovascular risk remains to be investigated.
Deep molecular response in patients with chronic phase chronic myeloid leukemia treated with the plasminogen activator inhibitor-1 inhibitor TM5614 combined with a tyrosine kinase inhibitor (2023)
Abstract
Background: We recently showed that pharmacological inhibition of plasminogen activator inhibitor-1 (PAI-1) activity, based on TM5614, increases cell motility and induces the detachment of hematopoietic stem cells from their niches. In this TM5614 phase II clinical trial, we investigated whether the combination of a PAI-1 inhibitor and tyrosine kinase inhibitors (TKIs) would induce a deep molecular response (DMR) in patients affected by chronic myeloid leukemia (CML) by quantifying BCR-ABL1 transcripts.
Methods: Patients with chronic phase CML treated with a stable daily dose of TKIs for at least 1 year and yielding a major molecular response (MMR) but not achieving MR4.5 were eligible for this study. After inclusion, patients began to receive TM5614 as well as a TKI. The primary objective was an evaluation of the cumulative incidence of patient progression from an MMR/MR4 to MR4.5 by 12 months.
Results: Thirty-three patients were enrolled in the study. The median age was 59.0 years and 58% were male. No Sokal high-risk patients were enrolled in this trial. The median TKI treatment duration was 4.8 years. At the start of this study, seven patients and 26 patients received imatinib and second-generation TKIs, respectively. The cumulative MR4.5 incidence by 12 months was 33.3% (95% confidence interval, 18.0%-51.8%). The cumulative MR4.5 spontaneous conversion over 12 months was estimated as 8% with TKIs alone based on historical controls. The halving time of BCR-ABL1 at 2 months was significantly shorter for patients who achieved an MR4.5 , by 12 months than for the other patients (cutoff value: 48 days; sensitivity: 0.80; specificity: 0.91; ROC-AUC: 0.83). During this study, bleeding events and abnormal coagulation related to the drug were not reported, and TM5614 was found to be highly safe.
Conclusion: TM5614 combined with TKI was well tolerated and induced MR4.5 in more patients than stand-alone TKI treatment.
Previous studies have shown that other approaches such as IFN-A/peg-IFN-A,28,29 a BCL-2 inhibitor (venetoclax), a JAK2 inhibitor (ruxolitinib), a BTK inhibitor, a PPAR-γ (pioglitazone),30 and a PD-1 antibody in combination with TKI, can be applied to eliminate CML stem cells in patients with CML not achieving DMR with TKI monotherapy.31,32
All patients started daily TM5614 at a dose of 150mg/day, and this was increased to 180 mg/day after 8 weeks for 18 patients (54.5%) because those patients did not report any adverse events (AEs) or abnormal laboratory results and showed drug tolerance.
This phase II clinical trial by Takahashi et al. (2023) investigated whether combining a plasminogen activator inhibitor-1 (PAI-1) inhibitor called TM5614 with tyrosine kinase inhibitors (TKIs) could induce a deep molecular response (DMR), defined as MR4.5 (BCR-ABL1 ≤0.0032%), in patients with chronic myeloid leukemia (CML) who had not achieved this level of response with TKI therapy alone.
33 patients with chronic phase CML were enrolled. All had been on stable TKI therapy for over 1 year and had achieved major molecular response (MMR; BCR-ABL1 ≤0.1%) but not MR4.5.
7 patients were on imatinib and 26 were on 2nd generation TKIs dasatinib, nilotinib or bosutinib. Median duration of prior TKI therapy was 4.8 years.
Patients began TM5614 150 mg/day in addition to their TKI. Dose was increased to 180 mg/day in 18 patients after 8 weeks.
Primary endpoint was rate of MR4.5 by 12 months.
11 patients (33.3%) achieved MR4.5 by 12 months. Median time to MR4.5 was 8 weeks.
Historical MR4.5 rate with TKIs alone is estimated around 8% per year, so combination appears much more effective.
TM5614 levels were stable over time. No evidence of accumulation.
All patients had some degree of BCR-ABL1 reduction during the study. Patients achieving MR4.5 had larger reductions, particularly by 8 weeks.
Median halving time of BCR-ABL1 at 8 weeks was significantly shorter in patients achieving MR4.5 (5.3 vs 14.1 weeks).
Halving time <6.9 weeks at 8 weeks had sensitivity 80% and specificity 91% for predicting MR4.5 by 48 weeks.
Cumulative incidence of MR4.5 by 48 weeks was 80% for patients with halving time <6.9 weeks at 8 weeks vs only 9% for those with longer halving time.
10 of 11 patients maintained MR4.5 for median 266 days after stopping TM5614.
Multivariate analysis found baseline BCR-ABL1 level (MR4 vs MMR) only factor predicting MR4.5.
No major safety issues. AEs generally mild and not clearly related to TM5614.
In summary, the key findings of this phase II trial by Takahashi et al. (2023) were:
Combining the PAI-1 inhibitor TM5614 with TKIs resulted in significantly higher rates of deep molecular response (33% MR4.5 at 1 year) compared to historical rates with TKIs alone (~8% per year).
Faster reduction of BCR-ABL1, particularly halving time <6.9 weeks at 8 weeks, strongly predicted achievement of MR4.5 by 48 weeks.
Most patients achieving MR4.5 maintained response for 8+ months after stopping TM5614.
Baseline BCR-ABL1 level (MR4 vs MMR) was only independent predictor of MR4.5 in multivariate analysis.
No major safety issues were identified with the addition of TM5614 to TKIs.
The authors conclude that adding TM5614 to TKIs is a promising approach to induce high rates of deep molecular response in patients with CML not achieving this level of response after years of TKI therapy alone. If confirmed in larger randomized trials, this combination strategy could increase the number of CML patients eligible for treatment free remission.
Impacts of Bacillus subtilis var. natto on the lifespan and stress resistance of Caenorhabditis elegans (ref)
A null mutation in SERPINE1 protects against biological aging in humans (2017)
TM5614, an Inhibitor of Plasminogen Activator Inhibitor-1, Exerts an Antitumor Effect on Chronic Myeloid Leukemia (2022)
The authors established K562 chronic myeloid leukemia (CML) cell clones that stably overexpress plasminogen activator inhibitor-1 (PAI-1).
Treatment of these cells with the PAI-1 inhibitor TM5614 significantly reduced cell proliferation and induced apoptosis.
TM5614 decreased PAI-1 levels and activity and increased activity of the protease Furin. Furin activates membrane-type matrix metalloprotease-1 (MT1-MMP), which detaches CML stem cells from their niche.
TM5614 also increased levels of NOTCH1 intracellular domain (NICD) and NOTCH target genes HES1 and HEY1.
Overexpression of Furin or NICD in the PAI-1-overexpressing cells mimicked the anti-proliferative and pro-apoptotic effects of TM5614.
The authors conclude that PAI-1 inhibition by TM5614 exerts anti-tumor effects in CML cells by modulating the Furin/NICD pathway.
They suggest PAI-1 blockade could be a therapeutic strategy for CML, based on the direct anti-tumor effects shown in this study as well as previous work showing PAI-1 inhibition detaches CML stem cells from their niche.
In summary, this study demonstrated novel anti-leukemic effects of the PAI-1 inhibitor TM5614 in CML cells, likely mediated through activation of Furin and NOTCH1 signaling. The results support further investigation of PAI-1 inhibition as a therapeutic approach for CML.
A phase i and pharmacodynamic study of AT9283, a small-molecule inhibitor of aurora kinases in patients with relapsed/refractory leukemia or myelofibrosis (2014)
Blockade of plasminogen activator inhibitor-1 empties bone marrow niche sufficient for donor hematopoietic stem cell engraftment without myeloablative conditioning (2019)
Upon hematopoietic stem cell transplantation (HSCT), the availability of recipients' niches in the bone marrow (BM) is one of the factors that influence donor HSC engraftment and hematopoietic reconstitution. Therefore, myeloablative conditioning, such as irradiation and/or chemotherapy, which creates empty niches in the recipients' BM, is required for the success of HSCT. However, the conventional myeloablation causes extensive damages to the patients' BM, which results in the treatment-induced severe complications and even mortality. Thus, alternative and mild conditioning could fulfill the need for safer HSCT-based therapies for hematological and nonhematological disorders. Recently, we have demonstrated that pharmacological inhibition of plasminogen activator inhibitor-1 (PAI-1) activity increases cellular motility and cause detachment of HSCs from the niches. In this study, we performed HSCT using a PAI-1 inhibitor without any myeloablative conditioning. Donor HSCs were transplanted to recipient mice that were pretreated with saline or a PAI-1 inhibitor. Saline pretreated nonmyeloablative recipients showed no engraftment. In contrast, donor cell engraftment was detected in the PAI-1 inhibitor pretreated recipients. Multilineage differentiation, including lymphoid and myeloid cells, was observed in the PAI-1 inhibitor pretreated recipients. Donor-derived cells that exhibited multilineage reconstitution as well as the existence of stem/progenitor cells were detected in the secondary recipients, confirming the maintenance of donor HSCs in the BM of PAI-1 inhibitor pretreated primary recipients. The results indicate that the PAI-1 blockade vacates functional niches in the recipients’ BM, which allows the engraftment of long-term multilineage HSCs without myeloablative conditioning.
Nattokinase: A Promising Alternative in Prevention and Treatment of Cardiovascular Diseases (2018)
In vitro studies show that nattokinase decreases clot formation by cleaving and inactivating the plasminogen activator inhibitor (PAI) via proteolysis at P1-P1’ peptide bond. PAI is a key inhibitor of tissue plasminogen activator (tPA) that converts plasminogen to plasmin. PAI inactivation allows for greater tPA activity and increased lysis of clots (10) (11). In the absence of PAI, nattokinase affects direct proteolysis of fibrin; however, this effect is less than the protelysis achieved by the PAI-mediated pathway (2). The fibrinolytic activity of nattokinase is estimated to be four-fold that of plasmin (12).
It is now known that NK not only degrades fibrin directly and effectively but also increases the release of tPA with a subsequent increase in the formation of plasmin.5,17,26–28 Plasminogen activator inhibitor 1 (PAI-1) is the primary inhibitor of tPA and regulates fibrinolytic activity in the fibrinolytic cascade.29 In a study investigating the mechanism by which NK exerted its fibrinolytic effect, NK enhanced fibrinolysis through cleavage and inactivation of PAI-1.4 In this study, NK was shown to cleave active recombinant prokaryotic PAI-1 into low-molecular-weight fragments as well as enhance tissue-type plasminogen activator–induced fibrin clot lysis. The enhanced fibrinolytic activity observed in the absence of PAI-1 appeared to be induced through direct fibrin dissolution by NK.4 Nattokinase also enhanced the production of clot-dissolving agents such as urokinase through the conversion of prourokinase to urokinase.5,19 Furthermore, NK was shown to be capable of blocking thromboxane formation resulting in an inhibition of platelet aggregation without producing the side effect of bleeding.12 Thus, NK was found to be a potent antithrombotic agent, and, by reducing thrombus formation, was able to slow the progression of plaque formation and reverse evolving atherosclerotic lesions.10
Herb Lab Interactions
The profibrinolytic enzyme subtilisin NAT purified from Bacillus subtilis cleaves and inactivates plasminogen activator inhibitor type 1 (2001)
High plasma levels of plasminogen activator inhibitor 1 (PAI-1) in polycythemia vera and essential thrombocythemia are associated with thrombosis (1994)
We attempted to determine if a hypercoagulability state exists in patients with polycythemia vera (PV) and essential thrombocythemia (ET). We studied the hematocrit level, platelet count, use of any antiaggregant drugs, thrombotic or bleeding accidents and plasma levels of antithrombin III, protein C, total protein S, free protein S, vWF:Ag (Von Willebrand's factor related antigen), thrombin-antithrombin complexes, D-dimer, fibrinolytic activity, tissue plasminogen activator, plasminogen and PAI-1 in 33 patients (19 with ET and 14 with PV). PAI-1 plasma concentration was significantly higher in, both ET and PV patients than in the control group, and were higher in those patients with previous thrombotic episodes than in asymptomatic patients or with previous bleeding episodes. Increasing age was associated to more thrombotic episodes while younger patients presented with more hemorrhagic complications. A linear correlation between platelet count and PAI-1 levels in PV patients (r = 0.44, p < 0.05) and ET patients (r = 0.30, p < 0.05) was found. Fibrinolytic activity in patients with ET was significantly decreased when compared to the control group. A hypofibrinolytic state could be an additional factor which could be used as a predictive index of the thrombotic or bleeding tendency in each patient.
A Narrative Review on Plasminogen Activator Inhibitor-1 and Its (Patho)Physiological Role: To Target or Not to Target? (2021)
Plasminogen activator inhibitor-1 (PAI-1) is the main physiological inhibitor of plasminogen activators (PAs) and is therefore an important inhibitor of the plasminogen/plasmin system. Being the fast-acting inhibitor of tissue-type PA (tPA), PAI-1 primarily attenuates fibrinolysis. Through inhibition of urokinase-type PA (uPA) and interaction with biological ligands such as vitronectin and cell-surface receptors, the function of PAI-1 extends to pericellular proteolysis, tissue remodeling and other processes including cell migration. This review aims at providing a general overview of the properties of PAI-1 and the role it plays in many biological processes and touches upon the possible use of PAI-1 inhibitors as therapeutics.
PAI-1 is synthesized by endothelial cells, hepatocytes, adipocytes, fibroblasts, smooth muscle cells, monocytes, macrophages, and platelets. Its expression and release are strongly regulated by growth factors, cytokines, hormones, glucose, endotoxin, etc. PAI-1 circulates in plasma or is stored in platelets. Plasma PAI-1 is mainly active while platelet PAI-1 is largely latent, but can be activated upon platelet stimulation.
PAI-1 is a serine protease inhibitor (serpin) with an exposed reactive center loop (RCL) that mimics the normal substrate of plasminogen activators. Upon binding to a plasminogen activator, PAI-1 can act as an inhibitor by inserting its cleaved RCL into its central β-sheet A, irreversibly distorting the plasminogen activator. Alternatively, PAI-1 can act as a substrate if the acyl-enzyme intermediate is hydrolyzed before RCL insertion and plasminogen activator distortion occur.
Active PAI-1 spontaneously converts to an inert latent form by inserting its RCL. The active form is stabilized ~2-fold by binding vitronectin. Small molecules, mutations, and conditions (pH, arginine, etc.) also affect PAI-1 stability.
Apart from inhibiting proteases, PAI-1 interacts with vitronectin (preventing cell interactions), and LDL receptor family members LRP1 and VLDLR (mediating cellular clearance). Through these interactions, PAI-1 is involved in various physiological and pathological processes independent of its antiprotease activity.
Elevated PAI-1 levels are linked with cardiovascular disease, metabolic disturbances, aging, cancer, tissue fibrosis, inflammation, and neurodegeneration. Specific contributions in these disease areas are:
Cardiovascular disease: By inhibiting fibrinolysis, PAI-1 promotes a prothrombotic state associated with myocardial infarction, stroke, venous thrombosis, and atherosclerosis. However, some studies found no significant association after adjusting for confounding factors.
Metabolic disturbances: High PAI-1 levels correlate with obesity, insulin resistance, type 2 diabetes and metabolic syndrome. PAI-1 expression is induced by glucose, insulin, and free fatty acids. PAI-1 promotes inflammation in adipose tissue. Lowering PAI-1 levels ameliorates metabolic abnormalities.
Inflammation: PAI-1 facilitates early host defense but exacerbates uncontrolled inflammation in sepsis and COVID-19 by promoting microthrombi formation.
Cancer: Despite its antiprotease effects, PAI-1 demonstrates mostly pro-tumorigenic properties by inhibiting apoptosis and promoting angiogenesis and cell migration. However, its role depends on many factors and some cancers show no correlation between PAI-1 levels and prognosis.
Fibrosis: By inhibiting ECM degradation, excess PAI-1 promotes fibrosis in organs including skin, lung, kidney, and liver. In contrast, PAI-1 deficiency can also enhance cardiac fibrosis.
Neurodegeneration: PAI-1 promotes microglial migration but impairs fibrin clearance in multiple sclerosis. It also reduces amyloid-β clearance in Alzheimer's disease.
Aging: Cellular senescence increases PAI-1 expression. Lower PAI-1 levels (in Amish null allele carriers) associate with longer lifespan and healthier metabolism.
Multiple approaches have been used to develop PAI-1 inhibitors, including small molecules, peptides, antibodies, and antisense oligonucleotides. These inhibitors act by:
Blocking PAI-1/plasminogen activator interactions
Inducing PAI-1 substrate behavior
Accelerating conversion to latent/inactive PAI-1
Inhibiting PAI-1 binding to ligands like LRP1
The binding sites and mechanisms of action for different PAI-1 inhibitors have been elucidated using various techniques. Small molecules often bind the flexible joint region or the sB/sC pocket, inducing substrate behavior or blocking initial interactions with plasminogen activators, respectively. Peptides insert into the PAI-1 core beta-sheet A, either inducing substrate behavior or accelerating latency conversion depending on their position. Antibodies prevent PAI-1/plasminogen activator interactions or induce substrate behavior or latency conversion by binding diverse PAI-1 epitopes.
Although promising preclinical and early clinical data have been obtained, no PAI-1 inhibitors are yet approved for therapeutic use. However, the development of safe and effective PAI-1 modulators remains an active area of pharmaceutical research for treating PAI-1-associated diseases.
In conclusion, PAI-1 is an important regulator of proteolytic systems and physiological processes, but its elevated expression promotes disease progression in many pathologies. Therapeutic targeting of PAI-1 shows promise but further research is needed to develop approved inhibitors. Careful modulation rather than complete inhibition of PAI-1 may be required to obtain clinical benefits while avoiding potential safety risks.
Treatment related changes in antifibrinolytic activity in patients with polycythemia vera (2010)
Polycythemia vera (PV) is a clonal myeloproliferative disorder characterized by predominantly excessive erythrocyte production. During the course of the disease, bleeding or thrombosis may be observed. In PV patients, the influence of antifibrinolytic activities on development of thrombohemorrhagic complications remains to be elucidated. In the present study, alterations in antifibrinolytic activity of PV patients and the effects of treatments on these alterations were investigated. Newly diagnosed and therapy-naive 22 PV patients were included. Thrombomodulin (TM), plasmin-alpha 2-antiplasmin complex (PAP), plasminogen activator inhibitor-1 (PAI-1) and thrombin activable fibrinolysis inhibitor antigen (TAFIa) levels were measured in all individuals and after phlebotomy and 5-hydroxyurea (5-HU) therapy in PV patients. TM, PAP, PAI-1 and TAFIa values of the patient group were higher than those of the controls. After phlebotomy, no changes were detected in TM, PAI-1 and TAFIa values, but PAP values decreased. On the contrary, 5-HU treatment resulted in a marked decrease in TM, PAI-1, PAP and TAFIa levels. These findings suggested that the changes in antifibrinolytic activity and endothelial dysfunction might be contributed to formation of intravascular thrombosis in PV patients, even though not clinically overt. 5-HU in addition to phlebotomy affects antifibrinolytic activity and may have an influence on diminishing predisposition of thrombosis.
Tissue Plasminogen Activator (t-PA) and Plasminogen Activator Inhibitor-1 (PAI-1) in Essential Thrombocythemia and Polycythemia Vera and Thrombotic Complications (2006)
Thrombotic complications are frequent in patients with essential thrombocythemia (ET) and polycythemia vera (PV). While a thrombin- and tissue plasminogen activator (t-PA)-regulated release of t-PA and plasminogen activator inhibitor-1 (PAI-1), respectively, has been shown and an association between elevated t-PA and PAI-1 and thrombogenesis has been studied for several vascular diseases, few studies have concentrated on the correlation between t-PA and PAI-1 levels and thrombogenesis in these myeloproliferative disorders. Therefore, we investigated prothrombin fragment 1+2 (F1+2), as marker of thrombin generation, and t-PA and PAI-1 in patients with ET and PV and thrombosis. We included 44 patients, 22 ET (5 men and 17 women, mean age 55 years) and 22 PV (17 men and 5 women, mean age 60 years) who fulfilled PVSG. The mean duration of disease was 4.5 years. Most received cytoreduction, such as hydroxyurea (13 ET and 14 PV), interferon-alpha (2 ET), anagrelide hydrochloride (5 ET) or phlebotomy (8 PV). None of studied patients had thrombotic risk factors. Of 44 patients, 25 (12 ET and 13 PV) developed thrombosis, whereas 19 (10 ET and 9 PV) did not. Measurements were assayed by ELISA. All patients had increased F1+2 (2.8±2.8 nmol/L vs 0.7±0.2 nmol/L) (p=0.001). t-PA and PAI-1 were higher in those patients with previous thrombosis than in asymptomatic patients (130±62 ng/ml vs 89±69 ng/ml and 45±20 ng/ml vs 57±27 ng/ml, respectively) (p=0.043 and p=0.085, respectively). A positive correlation there was between F1+2 and t-PA and t-PA and PAI-1 (p<0.0001 and p<0.0001, respectively). These findings suggest that a disfibrinolysis may be in ET and PV and that t-PA and PAI-1 may be predictive additional factors of the thrombotic tendency of these patients.
Why these Amish live longer and healthier: an internal ‘fountain of youth’ (link)
The mutation was identified in a girl from the community who almost died at age 3 due to excessive bleeding after bumping her head. She was found to have a rare deficiency of PAI-1 (plasminogen activator inhibitor-1), a protein involved in blood clotting.
Further testing of her extended family tree, spanning over 10,000 individuals, identified 12 patients with the bleeding disorder who had two copies of the mutated PAI-1 gene. About 96 others were found to carry one copy of the mutated gene but did not have the bleeding disorder.
Those kindred (family members) with one copy of the mutated gene lived over 10% longer (to age 85 on average) than average life expectancy of 71 for Amish, and had 10% longer protective telomeres than those without the mutation.
Carriers of the mutation had 29% lower fasting insulin levels and were completely protected from diabetes, with 0% incidence compared to 7% incidence in non-carriers.
The mutation carriers also showed improvements in cardiovascular measures including blood pressure and blood vessel stiffness, indicative of more youthful, flexible arteries.
The researchers suggest the consistent anti-aging benefits across metabolic (insulin), cardiovascular, and cellular aging (telomeres) parameters indicate the PAI-1 mutation confers longevity by protecting against biological aging.
An oral drug called TM5614 that inhibits PAI-1 is being developed by Renascience and tested in clinical trials in Japan.
When tested in aging mice models, the PAI-1 inhibitor drug extended lifespan more than 3-fold compared to controls and protected lungs and blood vessels from aging-related damage.
The researchers propose that since PAI-1 is a biomarker of cell senescence and aging, inhibiting it may protect against aging on both the cellular and physiological level in humans as it appears to do in mice.
Phase 1 trials in Japan showed the PAI-1 inhibitor drug TM5614 to be safe in humans. Phase 2 trials are underway to see if it boosts blood cell counts in chemo patients by promoting stem cell migration from bone marrow.
An early topical formula of the drug grew hair in bald mice, so a version is being developed to treat male pattern baldness.
A Serpin With a Finger in Many PAIs: PAI-1's Central Function in Thromboinflammation and Cardiovascular Disease (2021)
Morrow et al. (2021) reviewed the central role of plasminogen activator inhibitor-1 (PAI-1) in thromboinflammation and cardiovascular disease. PAI-1 is an unstable serine protease inhibitor that is the principal inhibitor of tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA), regulating fibrinolysis.
Key points:
PAI-1 levels are elevated in cardiovascular diseases including myocardial infarction, stroke, deep vein thrombosis, and atherosclerosis. High PAI-1 tilts the hemostatic balance toward hypofibrinolysis and thrombotic complications.
Inflammation dramatically increases PAI-1 levels due to cytokines like IL-6, TNF-α, TGF-β. This links inflammation to hypofibrinolysis. High PAI-1 is seen in sepsis, COVID-19, infections.
PAI-1 is a critical feature of metabolic syndrome and its components like obesity, type 2 diabetes, and hypertension. It is considered a true component of metabolic syndrome.
Adipose tissue is a major source of elevated PAI-1 in obesity and metabolic syndrome. Visceral fat secretes more PAI-1 than subcutaneous fat.
PAI-1 levels strongly predict development of type 2 diabetes, even independent of obesity and insulin. PAI-1 may contribute to insulin resistance.
PAI-1 deficiency or inhibitors prevent hypertension, obesity, insulin resistance in animal models, indicating promising therapeutic potential.
Conclusions:
Elevated PAI-1 is conclusively linked to thrombosis through hypofibrinolysis in cardiovascular diseases.
Inflammation dramatically increases PAI-1, linking inflammatory states to thrombotic complications.
PAI-1 is central to metabolic syndrome pathophysiology, especially from adipose sources.
Strong evidence confirms PAI-1's role in obesity, diabetes, hypertension.
PAI-1 elevation is a common thread connecting thrombosis, inflammation, and metabolic syndrome.
More research on cellular sources of PAI-1 in specific diseases is needed.
Clinical trials of PAI-1 inhibitors show promising results for thrombosis, obesity, diabetes.
In summary, this comprehensive review clearly establishes PAI-1 as a central mediator connecting thrombosis, inflammation, and metabolic syndrome. Converging evidence from human studies and animal models demonstrates PAI-1's pivotal role in cardiovascular pathologies through hypofibrinolysis, as well as metabolic disorders like obesity and diabetes. PAI-1 represents a promising therapeutic target, as supported by preclinical studies. Further research should focus on testing PAI-1 inhibitors in clinical trials and elucidating disease-specific cellular sources and mechanisms.
Plasminogen Activator Inhibitor 1 (PAI-1) Activity (Labcorp)
Targeting of plasminogen activator inhibitor-1 activity promotes elimination of chronic myeloid leukemia stem cells (2021)
The study by Yahata et al. (2021) investigated the role of plasminogen activator inhibitor-1 (PAI-1) in protecting chronic myeloid leukemia (CML) stem cells from tyrosine kinase inhibitors (TKIs) like imatinib. The authors found that the TGF-β-PAI-1 axis was selectively activated in CML stem cells compared to normal hematopoietic stem/progenitor cells. Overexpression of intracellular PAI-1 (iPAI-1) in CML cells made them more resistant to imatinib-induced apoptosis in vitro and in vivo. Conversely, PAI-1 knockdown sensitized CML cells to imatinib. The authors suggest that iPAI-1 contributes directly to TKI resistance in CML by inhibiting apoptosis.
Treatment of CML mice with a PAI-1 inhibitor alone did not improve survival, but combining a PAI-1 inhibitor with imatinib significantly reduced CML cells in the bone marrow, decreased spleen size, and prolonged survival compared to imatinib alone. The combination therapy also overcame imatinib resistance in CML mice transplanted with PAI-1 overexpressing CML cells. Using a tetracycline-inducible CML mouse model, the authors again found that imatinib plus a PAI-1 inhibitor decreased BCR/ABL expression in CML stem cells and improved survival compared to imatinib alone.
Mechanistically, the authors showed that PAI-1 inhibition increased MT1-MMP-dependent motility of CML stem cells, altered expression of adhesion molecules like CD44 and VLA-4, and reduced interaction between CML stem cells and the protective bone marrow niche. An MT1-MMP neutralizing antibody abolished the therapeutic effects of PAI-1 inhibition. Serial transplantation experiments showed that combined imatinib and PAI-1 inhibitor treatment, but not imatinib alone, prevented recurrence of CML in secondary recipients, indicating elimination of CML stem cells. Adding an MT1-MMP antibody again blocked the benefit of PAI-1 inhibition.
In summary, key findings were:
TGF-β-PAI-1 signaling is selectively activated in CML stem cells and contributes to TKI resistance.
iPAI-1 protects CML cells from imatinib-induced apoptosis in vitro and in vivo.
PAI-1 inhibition sensitizes CML cells to imatinib in mouse models and improves survival.
The combination of imatinib and a PAI-1 inhibitor eliminates CML stem cells and prevents disease recurrence in transplant recipients.
PAI-1 inhibition acts by increasing MT1-MMP-dependent motility and disrupting the interaction of CML stem cells with the protective niche.
MT1-MMP is critical for the therapeutic benefit of PAI-1 inhibition in CML.
The authors conclude that the TGF-β-PAI-1 axis contributes directly to TKI resistance in CML stem cells by regulating apoptosis and retention in the niche. They suggest that selective PAI-1 inhibition represents a novel therapeutic approach to overcome TKI resistance by mobilizing CML stem cells from the niche and increasing their susceptibility to TKIs. This study provides a rationale for targeting PAI-1 in combination with TKIs like imatinib to eradicate CML stem cells and achieve sustained remission.
PAI-1 and the Metabolic Syndrome (2006)
The metabolic syndrome (MetS) is associated with overexpression of plasminogen activator inhibitor-1 (PAI-1). The mechanisms involved in inducing PAI-1 overexpression are complex and likely involve multiple factors acting simultaneously.
PAI-1 is considered a true component of the MetS. Plasma PAI-1 levels correlate with the severity of MetS, and improving insulin resistance through weight loss or medication decreases PAI-1 levels.
Possible mechanisms linking PAI-1 to MetS include:
Adipose tissue, especially visceral fat, is a major source of PAI-1. Macrophages infiltrating the adipose tissue during obesity may contribute significantly to PAI-1 production.
Inflammatory cytokines like TNF-alpha and TGF-beta stimulate PAI-1 production and are upregulated in obesity. They may mediate effects of insulin resistance on PAI-1.
Glucocorticoids like cortisol stimulate PAI-1 production. Local conversion of cortisone to cortisol by 11β-HSD1 in adipose tissue may drive PAI-1 overexpression.
Lipid disturbances like elevated insulin, proinsulin, free fatty acids, and VLDL can directly induce PAI-1 in cell cultures. However clinical correlations are not always observed.
Oxidative stress and hypoxia in expanded adipose tissue upregulate PAI-1.
Recent evidence suggests PAI-1 may contribute to obesity and insulin resistance, rather than just being a marker.
High baseline PAI-1 levels predict later development of diabetes, even after adjusting for obesity and insulin resistance.
PAI-1 inhibits insulin signaling pathways in cell cultures. PAI-1 deficiency improves adipocyte differentiation and insulin sensitivity.
Transgenic mice overexpressing PAI-1 have smaller adipocytes and impaired adipose expansion. Mice lacking PAI-1 are protected from obesity and metabolic dysfunction in some models.
Mechanisms may involve PAI-1 inhibition of insulin signaling, interaction with TGF-beta, or central nervous system effects via inhibition of tPA.
In summary, the MetS induces complex PAI-1 overexpression. In turn, PAI-1 may exacerbate obesity, insulin resistance, and cardiovascular complications. Developing PAI-1 inhibitors could be beneficial for managing both atherothrombosis and metabolic disturbances.
Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions (2010)
The study by Cesari et al. (2010) provides a comprehensive review of the role of plasminogen activator inhibitor-1 (PAI-1) in age-related subclinical and clinical conditions. PAI-1 is a member of the serpin superfamily and the principal inhibitor of tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA), which activate plasminogen and initiate fibrinolysis. The authors summarize evidence that PAI-1 levels increase with age and are associated with a prothrombotic state that contributes to cardiovascular and cerebrovascular risk in the elderly.
Key points from the review:
Aging leads to impaired fibrinolysis, with prolonged euglobulin lysis time and prothrombotic changes in coagulation factors like increased PAI-1 and decreased tPA. This likely contributes to the increased risk of thrombotic events like myocardial infarction and stroke in older adults.
PAI-1 is produced by many cell types including endothelial cells, adipocytes, hepatocytes, and platelets. It inhibits fibrin breakdown by tPA and uPA. Elevated PAI-1 suppresses fibrinolysis and promotes fibrin deposition in tissues.
PAI-1 levels show circadian variation, peaking in the morning when risk of acute thrombotic events is highest.
PAI-1 synthesis is induced by inflammatory cytokines like IL-6, TNF-α, TGF-β; metabolic factors like insulin, glucose, triglycerides; and hormones like glucocorticoids. Mental/physical stress also increases PAI-1.
A common PAI-1 gene variant (4G/5G) affects baseline PAI-1 levels, with 4G associated with higher levels, but other factors like BMI more strongly determine concentrations.
Obesity is linked to impaired fibrinolysis and elevated PAI-1, especially from visceral fat. Adipose tissue secretes cytokines that further increase PAI-1 production. Weight loss reduces PAI-1.
Inflammation stimulators like TNF-α and IL-6 increase PAI-1 levels. PAI-1 is considered an acute phase reactant. Systemic inflammation in aging likely contributes to higher PAI-1.
Metabolic syndrome features like insulin resistance, hyperglycemia, and dyslipidemia are tied to elevated PAI-1. PAI-1 is implicated in the increased CVD risk with metabolic syndrome.
In atherosclerosis, PAI-1 expression is increased in lesions and atheroma, contributing to plaque instability. But effects are complex, with some evidence of anti-migratory effects on smooth muscle cells.
Elevated PAI-1 predicts adverse outcomes in many conditions - MI, stroke, cancer, diabetes, pregnancy complications. It is considered a promising biomarker linking thrombosis, inflammation, metabolic disorders, and aging.
Lifestyle interventions like exercise and weight loss help reduce PAI-1 levels. Medications like PPAR-γ agonists, RAS inhibitors, statins may also have beneficial effects on the fibrinolytic system.
In summary, Cesari et al. present extensive evidence that age-related elevations in PAI-1 promote a prothrombotic state that likely contributes to increased cardiovascular risk and disease prevalence in older populations. Therapies targeting PAI-1 reduction may help restore fibrinolytic balance and improve outcomes. More research is still needed on the complex biology of PAI-1 in aging and disease.
PAI-1 Inhibition – Another Therapeutic Option for Cardiovascular Protection (2015)
The authors summarize evidence that PAI-1 levels correlate with risk factors like hypertension, obesity, insulin resistance, and diabetes. PAI-1 polymorphisms have also been linked to increased myocardial infarction risk in meta-analyses. PAI-1 is closely tied to the renin-angiotensin system, and risk factors for atherosclerosis modulate fibrinolysis components. PAI-1 promotes vascular smooth muscle cell proliferation and migration while inhibiting apoptosis, which can contribute to intimal hyperplasia and atherosclerosis. It is synthesized in tissues like adipose, liver, endothelium, and platelets. PAI-1 stabilizes clots by inhibiting tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA). When the balance shifts towards thrombosis, PAI-1 exposure increases platelet activation and unstable plaque formation.
In terms of therapeutic strategies, statins may increase tPA while decreasing PAI-1 via inhibition of HMG-CoA reductase and modulation of geranylgeranylated intermediates. However, clinical trials have shown mixed results on impacts on PAI-1 levels. Angiotensin converting enzyme (ACE) inhibitors can increase tPA release through bradykinin and decrease PAI-1 through angiotensin II inhibition. But clinical studies on impacts of ACE inhibitors or angiotensin receptor blockers on PAI-1 have also been inconsistent. Aldosterone may increase PAI-1 expression through the mineralocorticoid receptor, so antagonists could potentially reduce thrombotic events. Fibrates may attenuate PAI-1 expression by lowering triglycerides, but effects are not well established. 17β-estradiol can increase tPA but has risks like cancer. TNF-α inhibitors and weight loss/exercise may reduce PAI-1 levels. Specific small molecule PAI-1 antagonists have been tested in animal models but require more research to translate to humans.
In summary, the imbalance between tPA and PAI-1 can promote thrombosis and cardiovascular events. Current cardiovascular therapies may modulate PAI-1 levels and fibrinolysis indirectly. While inhibiting PAI-1 could be a promising approach, definitive clinical benefits have not yet been proven. More research is needed to establish whether targeting PAI-1 can improve outcomes in atherosclerosis and related complications. The key questions remain whether PAI-1 is a causal risk factor versus just a disease marker, and if lowering PAI-1 levels will alter cardiovascular risk independent of other effects.
Type 1 plasminogen activator inhibitor (PAI-1) in clear cell renal cell carcinoma (CCRCC) and its impact on angiogenesis, progression and patient survival after radical nephrectomy (2010)
A total of 172 consecutive patients with CCRCC treated with radical nephrectomy were enrolled in the study. The expression of PAI-1, TSP-1 and factor VIII were analysed on formalin-fixed, paraffin-embedded tissues without knowledge of the clinical outcome. Ten cases, where PAI-1 immunohistochemistry was not possible due to technical problems and lack of material, were excluded. Sixty-nine patients (43%) died of RCC, while 47 patients (29%) died of other diseases. Median follow-up was 13.8 years for the surviving 46 patients (28%).
Nine percent of the tumours showed PAI-1 positivity. High expression of PAI-1 was significantly inversely correlated with TSP-1 (p = 0.046) and directly with advanced stage (p = 0.008), high NG (3+4) (p = 0.002), tumour size (p = 0.011), microvessel density (p = 0.049) and disease progression (p = 0.002). In univariate analysis PAI-1 was a significant prognosticator of cancer-specific survival (CSS) (p < 0.001). Multivariate analysis revealed that TNM stage (p < 0.001), PAI-1 (p = 0.020), TSP-1 (p < 0.001) and MVD (p = 0.007) were independent predictors of CSS.
PAI-1 was found to be an independently significant prognosticator of CSS and a promoter of tumour angiogenesis, aggressiveness and progression in CCRCC.
Pivotal role of PAI-1 in a murine model of hepatic vein thrombosis (2006)
Hepatic veno-occlusive disease (VOD) is a common complication of high-dose chemotherapy associated with bone marrow transplantation. While the pathogenesis of VOD is uncertain, plasminogen activator inhibitor-1 (PAI-1) has emerged as a diagnostic marker and predictor of VOD in humans. In this study, we investigated the role of PAI-1 in a murine model of VOD produced by long-term nitric oxide synthase inhibition using L-NAME. After 6 weeks, wild-type (WT) mice developed extensive fibrinoid hepatic venous thrombi and biochemical evidence of hepatic injury and dysfunction. In contrast, PAI-1–deficient mice were largely protected from the development of hepatic vein thrombosis. Furthermore, WT mice that received tiplaxtinin, an antagonist of PAI-1, were effectively protected from L-NAME–induced thrombosis. Taken together, these data indicate that NO and PAI-1 play pivotal and antagonistic roles in hepatic vein thrombosis and that PAI-1 is a potential target in the prevention and treatment of VOD in humans.
Tiplaxtinin impairs nutritionally induced obesity in mice (2006)
To investigate the effect of tiplaxtinin, designed as a synthetic inhibitor of plasminogen activator inhibitor-1 (PAI-1), on obesity, male C57Bl/6 mice (13–14 weeks old) were kept on a high-fat diet (20.1 kJ/g) for four weeks without or with addition of tiplaxtinin (PAI-039) at a dose of 2 mg/g food. At the time of sacrifice, body weights were significantly lower in the inhibitor-treated mice (p < 0.0005). The weights of the isolated subcutaneous and gonadal fat deposits were also significantly lower (both p < 0.0005), associated with adipocyte hypotrophy. Inhibitor-treated adipose tissues displayed similar blood vessel size, but a higher blood vessel density. Fasting glucose and insulin levels, as well as glucose-tolerance tests were not significantly affected by the inhibitor treatment, whereas plasma triglyceride levels were significantly reduced (p = 0.02) and LDL-cholesterol levels significantly enhanced (p = 0.0002). Insulin-tolerance tests revealed significantly lower glucose levels at the end of the test in the inhibitor treated mice (p = 0.03). Thus, in this model of diet-in-duced obesity in mice administration of tiplaxtinin resulted in impaired adipose tissue development.
Here are some examples of PAI-1 inhibitor drugs
Tiplaxtinin (PAI-039) - One of the furthest along in development. Phase 2 trials found it was generally well tolerated.
PAI-749 - Also known as tifacogin, this is a recombinant form of the natural PAI-1 inhibitor protein TFPI. Studies are still early.
TM5275 - A small molecule PAI-1 inhibitor. Phase 1 trials found no major safety concerns.
S35225 - Developed by Sanofi-Aventis, this PAI-1 inhibitor was being studied for diabetic nephropathy but development was discontinued.
WAY-140312 - Also from Sanofi-Aventis, this one reached phase 1 trials but was discontinued.
ZK4044 - An oral PAI-1 inhibitor from Schering AG that made it to phase 1 testing but was not pursued further.
ETC-642 - Developed by Esperion Therapeutics, this oral small molecule reached phase 1 testing before being discontinued.
So in summary, the furthest along are Tiplaxtinin, Tifacogin, and TM5275. But none have advanced past phase 2 trials yet. Research is still ongoing to develop safer and more effective PAI-1 inhibiting drugs.
Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice (2012)
Plasminogen activator inhibitor-1 (PAI-1), an endogenous inhibitor of a major fibrinolytic factor, tissue-type plasminogen activator, can both promote and inhibit angiogenesis. However, the physiologic role and the precise mechanisms underlying the angiogenic effects of PAI-1 remain unclear. In the present study, we report that pharmacologic inhibition of PAI-1 promoted angiogenesis and prevented tissue necrosis in a mouse model of hind-limb ischemia. Improved tissue regeneration was due to an expansion of circulating and tissue-resident granulocyte-1 marker (Gr-1+) neutrophils and to increased release of the angiogenic factor VEGF-A, the hematopoietic growth factor kit ligand, and G-CSF. Immunohistochemical analysis indicated increased amounts of fibroblast growth factor-2 (FGF-2) in ischemic gastrocnemius muscle tissues of PAI-1 inhibitor-treated animals. Ab neutralization and genetic knockout studies indicated that both the improved tissue regeneration and the increase in circulating and ischemic tissue-resident Gr-1+ neutrophils depended on the activation of tissue-type plasminogen activator and matrix metalloproteinase-9 and on VEGF-A and FGF-2. These results suggest that pharmacologic PAI-1 inhibition activates the proangiogenic FGF-2 and VEGF-A pathways, which orchestrates neutrophil-driven angiogenesis and induces cell-driven revascularization and is therefore a potential therapy for ischemic diseases.
Novel oral plasminogen activator inhibitor-1 inhibitor TM5275 attenuates hepatic fibrosis under metabolic syndrome via suppression of activated hepatic stellate cells in rats
An orally bioavailable small molecule inhibitor of plasminogen activator inhibitor-1 (PAI-1) is currently being clinically assessed as a novel antithrombotic agent. Although PAI-1 is known to serve a key role in the pathogenesis of metabolic syndrome (MetS) including nonalcoholic steatohepatitis (NASH), the pharmacological action of an oral PAI-1 inhibitor against the development of MetS-related liver fibrosis remains unclear. The current study was designed to explicate the effect of TM5275, an oral PAI-1 inhibitor, on MetS-related hepatic fibrogenesis. The in vivo antifibrotic effect of orally administered TM5275 was investigated in two different rat MetS models. Fischer 344 rats received a choline-deficient L-amino-acid-defined diet for 12 weeks to induce steatohepatitis with development of severe hepatic fibrosis. Otsuka Long-Evans Tokushima Fatty rats, used to model congenital diabetes, underwent intraperitoneal injection of porcine serum for 6 weeks to induce hepatic fibrosis under diabetic conditions. In each experimental model, TM5275 markedly ameliorated the development of hepatic fibrosis and suppressed the proliferation of activated hepatic stellate cells (HSCs). Additionally, the hepatic production of tumor growth factor (TGF)-β1 and total collagen was suppressed. In vitro assays revealed that TGF-β1 stimulated the upregulation of Serpine1 mRNA expression, which was inhibited by TM5275 treatment in cultured HSC-T6 cells, a rat HSC cell line. Furthermore, TM5275 substantially attenuated the TGF-β1-stimulated proliferative and fibrogenic activity of HSCs by inhibiting AKT phosphorylation. Collectively, TM5275 demonstrated an antifibrotic effect on liver fibrosis in different rat MetS models, suppressing TGF-β1-induced HSC proliferation and collagen synthesis. Thus, PAI-1 inhibitors may serve as effective future therapeutic agents against NASH-based hepatic fibrosis.
Fibrinolytic response to interferon-alpha in healthy human subjects (1996)
Interferons (IFNs) are used for a variety of disorders. It has been postulated that part of the effects of IFN may be mediated by IFN-induced modulation of endothelial cells. Since the principal activating and inhibiting factors of the fibrinolytic system are synthesized and stored in endothelial cells, we have studied the effects on fibrinolysis and coagulation of the administration of recombinant IFN-alpha (5 x 10(6) U/m2) to healthy human subjects (n = 8) in a randomized controlled cross-over study. IFN-alpha significantly increased plasma levels of tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). Simultaneously, plasma levels of the inhibitor of plasminogen activation, PAI-1, sharply increased. The net effect on plasma plasminogen activator activity (PA-activity) was a modest increase to 116% of baseline, however without a significant effect on plasmin generation, as reflected by plasma levels of plasmin-alpha 2-antiplasmin complexes. IFN-alpha had no effect on the plasma levels of thrombin-antithrombin III (TAT) complexes. We conclude that despite considerable effects on endothelial cells, IFN-alpha does not significantly alter the coagulant-fibrinolytic balance, although the occurrence of such changes under pathological circumstances is not excluded.
Effects of Pegylated Interferon α on Fibrinolytic Parameters in Patients With Chronic Hepatitis C
Interferon (IFN) interacts with endothelial cells and modulates the functions of these cells. In our study, we aimed to determine the effects of treatment with pegylated IFN-α (peg-IFN-α) on fibrinolytic parameters in patients with chronic hepatitis C. Fifteen patients with chronic hepatitis C were treated with peg-IFN-α once per week plus daily oral ribavirin. Euglobulin lysis time (ELT), plasma levels of d-dimer, tissue-type plasminogen activator (t-PA), plasminogen activator inhibitor 1 (PAI-1), and thrombin activatable fibrinolysis inhibitor (TAFI) were determined before treatment, 2 weeks, 1 month, and 3 months after the initiation of the treatment. Plasma levels of t-PA increased significantly 1 month and 3 months after the treatment (P < .05). The PAI-1 and TAFI levels in 2 weeks, 1 month and 3 months after treatment were not statistically different as compared with pretreatment levels (P > .05) No significant difference in plasma d-dimer levels was observed during peg-IFN-α treatment (P > .05). There was a significant decrease in ELT 1 month and 3 months after the treatment (P < .05). Our results indicated that treatment with peg-IFN-α may be associated with enhanced fibrinolysis,
Plasminogen Activator Inhibitor-1 Levels in Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis
Background and aims: Several studies have investigated the role of multiple proteins in nonalcoholic fatty liver disease (NAFLD); one that has recently gained attention is plasminogen activator inhibitor-1 (PAI-1). However, studies evaluating PAI-1 levels in NAFLD demonstrated conflicting results. Our objective was to understand the role of PAI-1 in NAFLD more clearly by carrying out a systematic review and meta-analysis.
Methods: We gathered evidence by performing a systematic search on PubMed, EMBASE, and Cochrane Library, through using a predefined search string. The included studies diagnosed NAFLD through either liver biopsy, ultrasonography, computed tomography (CT), magnetic resonance spectroscopy, or using one of the latter methods with blood parameters. Studies had to fulfill predefined inclusion and exclusion criteria. To assess the quality of the studies included, we used the NHLBI quality assessment tools. The main summary outcome was the mean difference (MD) in serum PAI-1 levels reported as ng/mL Results: 33 articles involving 10,840 subjects fulfilled our inclusion criteria and were systematically reviewed. 11 studies were included in our meta-analyses. We found a significant MD in PAI-1 levels in NAFLD patients vs. controls [17.147 (95%CI: 7.720-26.574)]. Moreover, subgroup analysis evaluating PAI-1 levels in biopsy- proven NAFLD vs. controls remained significant [24.086 (95%CI: 3.812-44.361)], as well as in CT-diagnosed NAFLD [15.523 (95%CI: 7.163-23.883)]. However, no significant MD in PAI-1 levels was found in ultrasound- diagnosed NAFLD patients vs. controls [10.394 (95%CI: -13.335-34.123)]. No significant MD in PAI-1 levels in NASH patients vs. controls was observed [26.835 (95%CI: -0.879-54.549)].
Conclusions: In summary, elevated serum PAI-1 levels are associated with adult NAFLD (biopsy-proven and CT-diagnosed). However, no significant difference was found in ultrasound-diagnosed NAFLD and NASH patients. Nonetheless, the included studies have methodological variance, dictating that the obtained results should be carefully interpreted.
Inhibition of Plasminogen Activator Inhibitor 1 Attenuates Hepatic Steatosis but Does Not Prevent Progressive Nonalcoholic Steatohepatitis in Mice
Plasminogen activator inhibitor 1 (PAI-1), an essential regulator of fibrinolysis, is increasingly implicated in the pathogenesis of metabolic disorders, such as obesity and nonalcoholic fatty liver disease (NAFLD). Pharmacologic inhibition of PAI-1 is emerging as a highly promising therapeutic strategy for obesity and its sequelae. Given the well-established profibrotic function of PAI-1, we considered whether PAI-1 may serve as a target for antifibrotic therapy in nonalcoholic steatohepatitis (NASH). We therefore determined the effect of genetic Pai-1 deletion and pharmacologic PAI-1 inhibition on the development of NASH-related fibrosis in mice. Pai-1 knockout (Pai-1 -/-) and wild-type control (Pai-1 +/+) mice were fed a high-fat/high-cholesterol high-sugar (HFHS) diet or a methionine- and choline-deficient (MCD) diet to induce steatohepatitis with fibrosis. PAI-1 was pharmacologically inhibited using the small molecule inhibitor TM5441 in wild-type C57BL/6 mice fed an HFHS or MCD diet. Either genetic deletion of Pai-1 or pharmacologic inhibition of PAI-1 attenuated MCD diet-induced hepatic steatosis but did not prevent hepatic inflammation or fibrosis. Targeted inhibition of PAI-1 conferred transient protection from HFHS diet-induced obesity and hepatic steatosis, an effect that was lost with prolonged exposure to the obesigenic diet. Neither genetic deletion of Pai-1 nor pharmacologic inhibition of PAI-1 prevented HFHS diet-induced hepatic inflammation or fibrosis. Conclusion: Pai-1 regulates hepatic lipid accumulation but does not promote NASH progression. The PAI-1 inhibitor TM5441 effectively attenuates diet-induced obesity and hepatic steatosis but does not prevent NASH-related fibrosis in mice.
Small molecules inhibitors of plasminogen activator inhibitor-1 – An overview (pdf)
Here is a 2000 word summary of the key points and conclusions from the mini-review by Rouch et al. (2015):
Rouch et al. (2015) provide an overview of small molecule inhibitors targeting plasminogen activator inhibitor-1 (PAI-1), which plays a key role in regulating fibrinolysis and is implicated in many pathological conditions. PAI-1 inhibits the serine proteases tissue-type plasminogen activator (tPA) and urokinase plasminogen activator (uPA), thereby inhibiting fibrin clot breakdown. Due to its instability, native active PAI-1 has not been crystallized, hampering rational inhibitor design.
Peptides and pseudo-peptides mimicking the reactive center loop (RCL) of PAI-1 can induce latency transition. A 14-residue RCL peptide accelerated fibrinolysis in vitro (Eitzman et al., 1995), while a pentapeptide corresponding to P14-P10 bound two sites on PAI-1 in co-crystals. Shorter hexapeptides showed differential effects, with P14-P9 acting as a PAI-1 substrate and P8-P3 promoting latency. Dimeric pseudo-peptides TM5001 and TM5007 inhibited PAI-1 in vitro (IC50 28-29 μM) and reduced thrombosis in rat models without prolonging bleeding (Izuhara et al., 2008). Analogs with bulky hydrophobic groups maintained potency. TM5275 with improved oral bioavailability inhibited PAI-1 (IC50 5.6 μM) and reduced thrombosis in rats and monkeys without affecting bleeding. The binding sites and mechanisms were unclear.
Diketopiperazine-based compounds were the first small molecule PAI-1 inhibitors. XR334 and XR330 from Streptomyces sp. inhibited PAI-1 in vitro (IC50 30-52 μM) and increased fibrinolysis in rats (Bryans et al., 1996). Analogs XR5118 and XR11211 showed improved potency (IC50 0.2-6.4 μM), but poor drug-like properties. Related flat heteroaromatic scaffolds also inhibited PAI-1 moderately (IC50 1-50 μM), with octanoyl sidechains conferring higher potency (Folkes et al., 2002). XR5967 prevented tumor cell migration and angiogenesis by inhibiting PAI-1. The proposed binding site was between β-sheet A and α-helix F.
The salicylindole Fendosal (HP129) inhibited PAI-1 (IC50 13-75 μM), inducing a substrate-like conformation (Gils et al., 2002). The indole derivative Tiplaxtinin (PAI-039) discovered by HTS (IC50 2.7 μM) showed efficacy in animal models of thrombosis after oral dosing, and reversibly bound near α-helices D and E. PAI-749 (Diaplasinin) also inhibited PAI-1 (IC50 157 nM) via dual mechanisms, but lacked efficacy in human plasma. The binding sites and mechanisms of indole inhibitors were clarified using crystal structures, mutagenesis and modeling.
The benzofuran WAY-140312 and benzothiophene S35225 inhibited PAI-1 in vitro and reduced thrombosis in rats without prolonged bleeding. S35225 directly inhibited PAI-1 in plasma, while WAY-140312 required conversion to latency. The binding sites were not reported.
Menthol-based derivatives showed potent inhibitory activity (IC50 380 nM-1.4 μM), but issues with solubility and pharmacokinetics. Piperazine analog 28 discovered by HTS (IC50 500 nM) showed good oral bioavailability in rats, but only moderate efficacy. Hybrids with Tiplaxtinin had sub-micromolar potency, but again moderate efficacy in vivo (Pandya et al., 2011).
Oxadiazolidinedione 32 identified by HTS inhibited PAI-1 (IC50 5.3 μM), while derivative 33 with a phenyl linker showed 15-fold better potency (IC50 390 nM) (Gopalsamy et al., 2004). Oxalamides 34-37 with carboxylic acids and trifluoromethyl groups inhibited PAI-1 (IC50 4.5-8.4 μM), although the binding site was unclear (Jain et al., 2008). The pyrrolin-2-one T-1776Na inhibited PAI-1 (IC50 7.9 μM) and reduced thrombosis in rats, while analogs 39-40 showed 10-fold higher potency but no further studies (Miyazaki et al., 2009).
Polyphenolic compounds exhibit highly potent and reversible PAI-1 inhibition. The natural products sideroxylonals A-C inhibited PAI-1 (IC50 3.3-5.3 μM), binding directly to PAI-1 (Neve et al., 1999). Gallic acid had modest activity (IC50 6.6 μM) while tannic acid and epigallocatechin-3,5-digallate showed exceptional potency (IC50 7-91 nM). Synthetic polyphenols CDE-082 and CDE-066 inhibited PAI-1 in plasma and mice (Cale et al., 2010). Crystal structures show polyphenols bind allosterically between β-sheets and α-helices. However, poor drug-like properties remain an issue.
The antiflammatory flufenamic acid derivative AR-H029953XX inhibited PAI-1 (IC50 12 μM), proposed to bind near Phe114 and Arg115. The natural product embelin was crystallized with PAI-1 (PDB 3UT3), revealing a binding site overlapping with vitronectin. An analog showed improved potency (IC50 180 nM) (Chen et al., 2014). The azetidine AZ3976 uniquely inhibited latent PAI-1 by binding between helices D/F and β-strand 1A. However, vitronectin blocked this effect. No clinical candidates have yet emerged.
In summary, PAI-1 inhibitors have been intensely pursued for their therapeutic potential, with many chemical scaffolds exhibiting potent in vitro activity. However, issues such as poor pharmacokinetics, lack of efficacy in human systems or competition with stabilizing vitronectin have hampered translation to the clinic. Recent crystal structures have provided insight into binding sites and mechanisms of inhibitors, although rational design remains challenging. There is need for compounds that maintain efficacy in vivo and in human plasma toward developing effective PAI-1-targeted therapies.
Camellia sinensis Leaf Extract: Benefits, Uses, and Side Effects (link)
The plasminogen activator inhibitor-1 paradox in cancer: a mechanistic understanding (2019)
The review article examines the paradoxical pro-tumorigenic functions of plasminogen activator inhibitor-1 (PAI-1) in cancer progression and metastasis. PAI-1 is a member of the serine protease inhibitor (serpin) family and was originally expected to have anti-tumor effects by inhibiting urokinase plasminogen activator (uPA) activity. However, numerous clinical studies found that high tumor levels of PAI-1 are associated with poor prognosis in cancers like breast, ovarian, bladder, colon, and lung (Duffy et al, 1994; Janicke et al, 2001).
The complex and dynamic structure of PAI-1 explains its pleiotropic biological functions (Wind et al, 2002). PAI-1 exists in active, latent, and cleaved forms based on the configuration of its reactive center loop (RCL). It contains domains that bind uPA, vitronectin, and lipoprotein receptor proteins (LRP). Binding of uPA triggers cleavage of the RCL. Binding to vitronectin stabilizes active PAI-1. Binding to LRP triggers endocytosis. This multi-domain structure enables the diverse pro-tumorigenic functions of PAI-1.
Numerous clinical studies have validated uPA and PAI-1 in tumors as prognostic biomarkers guiding therapy decisions in cancers like breast and node-negative breast cancer (Janicke et al, 2001; Look et al, 2002). The prognostic value of the 4G/5G PAI-1 gene promoter polymorphism has also been examined, but results are not entirely clear (Lee et al, 2013).
Analyzing 3739 publications with text mining tools revealed PAI-1's pre-eminent roles in sustaining proliferative signaling, resisting cell death, angiogenesis, invasion/metastasis, and inflammation (Hanahan & Weinberg, 2011; Baker et al, 2017).
PAI-1 may stimulate cancer cell cycle progression through cyclin D3/Cdk4/6 (Giacoia et al, 2014). It modulates proliferation indirectly via effects on thrombin signaling, growth factor receptors, integrins, and extracellular matrix (McEachron et al, 2011; Mazzieri & Blasi, 2005). PAI-1 inhibits apoptosis by inhibiting caspase-3, reducing FasL shedding, and activating cell survival signaling via LRP-1 (Balsara & Ploplis, 2008; Fang et al, 2012).
PAI-1 promotes angiogenesis by inhibiting apoptosis and enabling migration in endothelial cells. It binds vitronectin, preventing endothelial adhesion, enabling migration toward fibronectin (Bajou et al, 2008). It increases fibrin deposition and release of angiogenic proteins like IL-8 (Qi et al, 1997; Olander et al, 1985).
In vitro, PAI-1 promotes migration by preventing cell adhesion and excessive ECM degradation (Liu et al, 1995). However, in vivo data on metastasis are conflicting, with evidence both for and against pro-metastatic effects (Maillard et al, 2008; Eitzman et al, 1996). More research on mechanisms is needed to clarify PAI-1's role.
PAI-1 contributes to cancer inflammation by increasing inflammatory cell recruitment and polarization towards pro-tumorigenic phenotypes (Kubala et al, 2018). It stimulates production of IL-8 and leukotriene B4 (Xu et al, 2009).
Despite the evidence for pro-tumorigenic effects, targeting PAI-1 has remained elusive. PAI-1 inhibitors have shown efficacy in some preclinical models but not others (Gorlatova et al, 2007; Mutoh et al, 2008). Challenges include insufficient pharmacokinetics, high therapeutic concentrations, and lack of activity against vitronectin-bound PAI-1 (Mashiko et al, 2015). Testing PAI-1 inhibitors in combination therapies and examining effects on the tumor microenvironment may reveal therapeutic potential.
In conclusion, this comprehensive review summarizes the complex structure of PAI-1 enabling its paradoxical pro-tumorigenic functions in cancer progression and metastasis. PAI-1 is a validated prognostic and therapy-guiding biomarker in cancers. It promotes proliferation, survival, angiogenesis, invasion/metastasis, and inflammation. However, challenges remain in therapeutically targeting PAI-1, warranting further research on mechanisms and combination therapies that could reveal its potential as a cancer therapy target.
Natural pai-1 inhibitors
Here are a few key points about natural PAI-1 inhibitors:
PAI-1 (plasminogen activator inhibitor-1) is a protein that inhibits fibrinolysis, the natural process of breaking down blood clots. High levels of PAI-1 are associated with increased risk of thrombosis and cardiovascular disease.
Some natural substances have been shown to inhibit PAI-1 activity, helping to promote fibrinolysis. These include curcumin (from turmeric), epigallocatechin gallate (EGCG from green tea), omega-3 fatty acids (from fish oil), and anthocyanins (from berries).
Curcumin has been widely studied and shown to significantly reduce PAI-1 levels. The mechanism is not entirely clear but may be related to curcumin's anti-inflammatory effects.
EGCG from green tea is also a potent PAI-1 inhibitor. Animal and lab studies show it lowers PAI-1 production and activity.
Omega-3s may inhibit PAI-1 by regulating gene expression. Population studies link higher omega-3 intake with lower PAI-1.
Berries high in anthocyanins like blueberries and cherries demonstrate PAI-1 lowering effects in studies. The anthocyanins likely regulate PAI-1 at the genetic level.
Fibrinolytic function after dietary supplementation with omega3 polyunsaturated fatty acids (1997)
Hypertension is associated with derangements in glucose and lipid metabolism. Increased levels of plasminogen activator inhibitor type 1 (PAI-1) are thought to potentiate the development of coronary events in this condition. Fish oil (omega3 polyunsaturated fatty acids [PUFAs]) have lipid-lowering effects, but the cardioprotective potential has been questioned because fish oil has been found to increase PAI-1 activity. This study was performed to determine the effects of omega3 PUFAs on the fibrinolytic function in hypertension. Seventy-eight persons with untreated hypertension were included in a 16-week, double-blind, randomized, controlled intervention study with 4 g/d of eicosapentaenoic and docosahexaenoic acids or corn oil placebo. Plasma PAI-1 activity, tissue plasminogen activator (tPA) activity, levels of fibrinogen and factor VII(c), and platelet count were measured before and after intervention (mean+/-SE). PAI-1 activity changed similarly in the fish oil and corn oil groups (1.8+/-1.0 U/mL versus 3.5+/-1.2 U/mL, P=.25), as did tPA (-0.02+/-0.02 IU/mL versus -0.13+/-0.03 IU/mL, P=.28), levels of factor VII(c) (6+/-5% versus 5+/-4%, P>.3), and platelet count (2+/-7x10(9)/L versus 3+/-5x10(9)/L, P>.3). None of these variables changed from pretreatment levels during fish oil intake. Fibrinogen levels increased significantly both during fish oil (0.6+/-0.1 g/L, P=.0001) and corn oil (0.4+/-0.1 g/L, P=.002) intake. There was no between-group difference (P>.3). In conclusion, a daily intake of 4 g omega3 PUFAs does not affect PAI-1 and tPA activity in persons with hypertension. A modest increase in fibrinogen levels was observed after both fish oil and corn oil intake.
Reduction in plasminogen activator inhibitor-1 (PAI-1) with omega-3 polyunsaturated fatty acid (PUFA) intake (1988)
Activity of plasminogen activator inhibitor-1 (PAI-1) in human blood correlates with thrombotic tendency and with serum triglyceride concentrations. Since intake of fish-derived omega-3 polyunsaturated fatty acids (PUFA) decreases serum triglycerides, we examined the effects of omega-3 PUFA maximum eicosapentaenoic acid (Max EPA) intake on PAI-1 levels in eight patients with coronary artery disease and in four normal subjects. After 4 weeks of Max-EPA intake by coronary artery disease patients, serum triglyceride concentrations and PAI-1 levels decreased 43 +/- 8% and 21 +/- 5%, respectively (both p less than or equal to 0.01) without any change in tissue plasminogen activator (TPA) levels. No changes were noted at 1 week of Max EPA intake in normal subjects, but at 3 weeks serum triglyceride concentrations and PAI-1 levels decreased 32 +/- 13% and 22 +/- 4%, respectively (p less than or equal to 0.01) without any change in tissue plasminogen activator (TPA) levels. No changes were noted at 1 week of Max EPA intake in normal subjects, but at 3 weeks serum triglyceride concentrations and PAI-1 levels decreased 32 +/- 13% and 22 +/- 4%, respectively (p less than or equal to 0.02) without change in TPA. The magnitude of reduction in triglycerides was dependent on the initial serum concentration (r = 0.68, p less than or equal to 0.01). In addition, decrease in PAI-1 levels correlated with reduction in serum triglycerides (r = 0.79, p less than or equal to 0.01). This study shows that omega-3 PUFA intake reduces PAI-1 levels without change in TPA antigen. These observations may relate to decrease in thrombotic activity upon consumption of large amounts of fish or fish-derived products.
Attenuation of the Synthesis of Plasminogen Activator Inhibitor Type 1 by Niacin
A Potential Link Between Lipid Lowering and Fibrinolysis (1995)
Background Plasminogen activator inhibitor type 1 (PAI-1), the primary physiological inhibitor of endogenous plasminogen activators, has been implicated as a potentiating factor in atherogenesis as well as in coronary thrombosis. We and others have observed attenuation of PAI-1 expression by gemfibrozil both in vivo and in vitro.
Methods and Results To determine whether other lipid-lowering agents with different mechanisms of action exert similar effects, we exposed Hep G2 cells, a highly differentiated human hepatoma cell line, to selected concentrations of niacin. Accumulation of PAI-1 protein, assayed with an ELISA, decreased in conditioned media by 72% in 48 hours in a specific, concentration-dependent fashion. Metabolic labeling experiments demonstrated a decrease in the rate of PAI-1 synthesis. Northern blot analysis demonstrated a preceding, parallel, and specific decrease in the concentration of PAI-1 mRNA. Niacin attenuated the increased PAI-1 synthesis induced by mediators released from thrombi as well. Thus, with 4.25 ng/mL transforming growth factor-β1, PAI-1 accumulation increased 4.5-fold in conditioned media in 48 hours. However, niacin attenuated the increase by 65%. Again, both the rate of PAI-1 synthesis and PAI-1 mRNA were reduced. The increased plasma PAI-1 activity and PAI-1 mRNA in liver induced by dexamethasone (0.8 mg IP) in vivo in rats were attenuated by 3 weeks of pretreatment with niacin.
Conclusions These results suggest that niacin, by decreasing PAI-1 expression, may potentiate fibrinolysis, thereby decreasing the stimulation of atherogenesis by clot-associated mitogens associated with microthrombi. Furthermore, the results imply that a pathogenetic link may exist between intracellular lipid metabolism and regulation of expression of fibrinolytic system components.
Supplements that increase fibronolysis
Here are some supplements that can help increase fibrinolysis or breakdown of blood clots:
Nattokinase - This enzyme extracted from natto fermented soybeans has been shown to enhance fibrinolytic activity and dissolve clots. Typical doses are 100-200 mg per day.
Lumbrokinase - Derived from earthworms, lumbrokinase is a fibrinolytic enzyme that may help prevent and break down clots. Usual dosage is 20-40 mg daily.
Fish Oil - The omega-3 fats EPA and DHA in fish oil supplements can reduce fibrinogen levels and platelet aggregation, promoting clot breakdown. Aim for 2-4 grams daily.
Vitamin E - Studies show vitamin E can stimulate release of PAI-1, an inhibitor of fibrinolysis. Dosage of 400-800 IU per day may help.
Ginger - Contains compounds called gingerols that exhibit antiplatelet activity and improve fibrinolytic activity. 500-2000 mg of ginger supplements daily.
Garlic - The ajoene compound in garlic has antithrombotic properties and can help prevent blood clots and enhance clot breakdown. Usual doses are 600-900 mg of garlic extract daily.
Bromelain - This enzyme from pineapple demonstrates fibrinolytic effects in studies. Typical doses are 200-400 mg 2-3 times per day.
Turmeric - Curcumin in turmeric has antiplatelet effects and can help restore blood flow and break down clots. 500-1000 mg daily is often recommended.
Supplements that may inhibit pai-1
Omega-3 Fatty Acids: Found in fish oil and certain plant oils, omega-3 fatty acids are known for their anti-inflammatory effects and may also help in reducing PAI-1 levels.
Green Tea Extract: The catechins in green tea, especially epigallocatechin gallate (EGCG), have been shown to have various health benefits and might influence PAI-1 levels.
Vitamin E: Some studies suggest that vitamin E, particularly in its natural form, might reduce PAI-1 production. However, its effectiveness can vary depending on individual health conditions and the presence of other antioxidants.
Curcumin: Found in turmeric, curcumin has anti-inflammatory properties and might affect PAI-1 levels, though its bioavailability is a concern and often requires enhanced formulation for effectiveness.
Resveratrol: This compound, found in grapes and berries, has been studied for various cardiovascular benefits and may influence PAI-1 levels.
Nattokinase: Derived from fermented soybeans, nattokinase is known for its fibrinolytic activity and might impact PAI-1, though more research is needed in this area.
Garlic Extract: Garlic and its extracts have been associated with various heart health benefits and may also affect PAI-1 levels.

How to track and optimize fibrinolysis? Is there a good blood marker to track? Any thoughts on what values are optimal?
Here are some ways to monitor and optimize fibrinolysis along with target values:
D-dimer blood test - This measures levels of fibrin degradation products, with higher levels indicating increased clot breakdown. Aim for a level <0.5 mcg/mL.
Fibrinogen blood test - Lower levels of this clotting factor suggest better fibrinolytic activity. Optimal range is 150-400 mg/dL.
PAI-1 (plasminogen activator inhibitor-1) - Lower circulating levels of this fibrinolysis inhibitor are better. Aim for <20 ng/mL.
tPA (tissue plasminogen activator) - Higher levels of this enzyme that activates plasminogen to break down clots are preferable. >10 ng/mL is a good target.
Euglobulin lysis time - This test measures time for a clot to lyse in a test tube. Faster times of 90-120 minutes indicate better fibrinolysis.
Thromboelastography - Provides information on clot formation and breakdown from a blood sample. An elevated lysis at 30 minutes parameter suggests increased fibrinolysis.

Comments