Horny Goat Weed (Icariin)
Inhibits JAK/STAT3 Signaling and Growth of Renal Cell Carcinoma.
inhibited tumor formation in various types of cancer cells, including breast cancer, leukemia, Burkitt lymphoma, liver cancer, stomach cancer, gallbladder cancer.
Shown to activate hepcidin pathways, through STAT3 activation.
Activates SIRT6 that is responsible for repairing DNA damage.
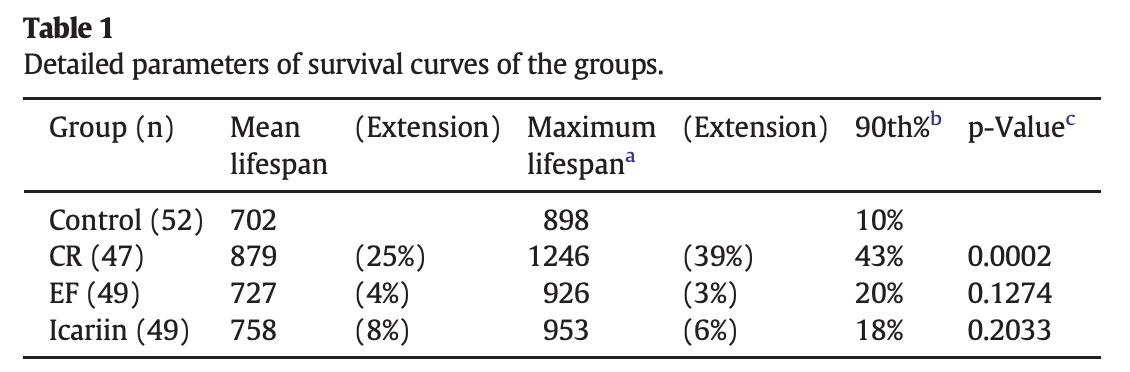
Extends mice lifespan by +8%.
May enhance libido. A weak PDE5 inhibitor. improved erectile function.
May produce Cytochrome P450 drug interactions in a dose-dependent manner.
Icariin, a natural flavonol glycoside, extends healthspan in mice

[It’s the PARPs and sirtuins (particularly SIRT6) that are responsible for repairing DNA damage. Best way to upregulate those is NMN and Icariin.]
So far, the compounds that can specifically regulate the activity of SIRT6 in CVDs are still limited. The Chinese herbal medicine, icariin, widely used in eastern countries to treat specific age-related diseases, including CVDs and the improvement of neurological function, has been proved to be an activator of SIRT6. In an in vitro cell model, it was discovered that 10–16–10–8mol/L icariin could effectively activate the expression of SIRT6 protein and delay cell senescence by inhibiting NF-κB signal transduction (inhibiting the expression of TNF-α, ICAM-1, IL-2, and IL-6). In the future, we need to supplement the clinical research of icariin in the treatment of CVDs (Chen et al., 2015).
Icariin Intervenes in Cardiac Inflammaging through Upregulation of SIRT6 Enzyme Activity and Inhibition of the NF-Kappa B Pathway
The aim of the study was to investigate the effect of icariin (ICA) on cardiac aging through its effects on the SIRT6 enzyme and on the NF-κB pathway. Investigating the effect of ICA on the enzymatic activity of histone deacetylase SIRT6 revealed a concentration of 10−8 mol/L ICA had a maximum activating effect on histone deacetylase SIRT6 enzymatic activity. Western analysis showed that ICA upregulated SIRT6 protein expression and downregulated NF-κB (p65) protein expression in animal tissues and cell models. ICA upregulated the expression of SIRT6 and had an inhibitory effect on NF-κB inflammatory signaling pathways as shown by decreasing mRNA levels of the NF-κB downstream target genes TNF-α, ICAM-1, IL-2, and IL-6. Those effects were mediated directly or indirectly by SIRT6. We provided evidence that inflammaging may involve a novel link between the effects of ICA on SIRT6 (a regulator of aging) and NF-κB (a regulator of inflammation).
Icaritin Inhibits JAK/STAT3 Signaling and Growth of Renal Cell Carcinoma, 2013
Signal transducer and activator of transcription-3 (STAT3) is critical for cancer progression by regulating tumor cell survival, proliferation, and angiogenesis. Herein, we investigated the regulation of STAT3 activation and the therapeutic effects of Icaritin, a prenyl flavonoid derivative from Epimedium Genus, in renal cell carcinoma (RCC). Icaritin showed significant anti-tumor activity in the human and mouse RCC cell lines, 786-O and Renca, respectively. Icaritin inhibited both constitutive and IL-6-induced phospho-STAT3 (STAT3Y705) and reduced the level of STAT3-regulated proteins Bcl-xL, Mcl-1, Survivin, and CyclinD1 in a dose-dependent manner. Icaritin also inhibited activation of Janus-activated kinase-2 (JAK2), while it showed minimal effects on the activation of other key signaling pathways, including AKT and MAPK. Expression of the constitutively active form of STAT3 blocked Icaritin-induced apoptosis, while siRNA directed against STAT3 potentiated apoptosis. Finally, Icaritin significantly blunted RCC tumor growth in vivo, reduced STAT3 activation, and inhibited Bcl-xL and Cyclin E, as well as VEGF expression in tumors, which was associated with reduced tumor angiogenesis. Overall, these results suggest that Icaritin strongly inhibits STAT3 activation and is a potentially effective therapeutic option for the treatment of renal cell carcinoma.
Cancer (link)
Icariin inhibited tumor formation in various types of cancer cells, including breast cancer, leukemia, Burkitt lymphoma, and liver cancer [72, 73, 74].
In stomach cancer cells, icariin suppressed tumor cell movement and invasion by inhibiting the Rac1-dependent VASP pathway, which is involved in cellular motility [75].
In another study, icariin suppressed the attachment and movement of stomach cancer cells by i
nactivation of protein kinase A (PKA), an enzyme known to promote cell production and survival [76].
Icariin inhibited gallbladder cancer cell production and induced cell death by suppressing NF-κB activity. The NF-kB pathway plays a key role in cancer cell survival by increasing anti-cell death factors survivin and Bc1 [72].
Icariin also triggered liver cancer cell death by activating the ROS/JNK-dependent mitochondrial pathway which plays a key role in initiating cell death [77].
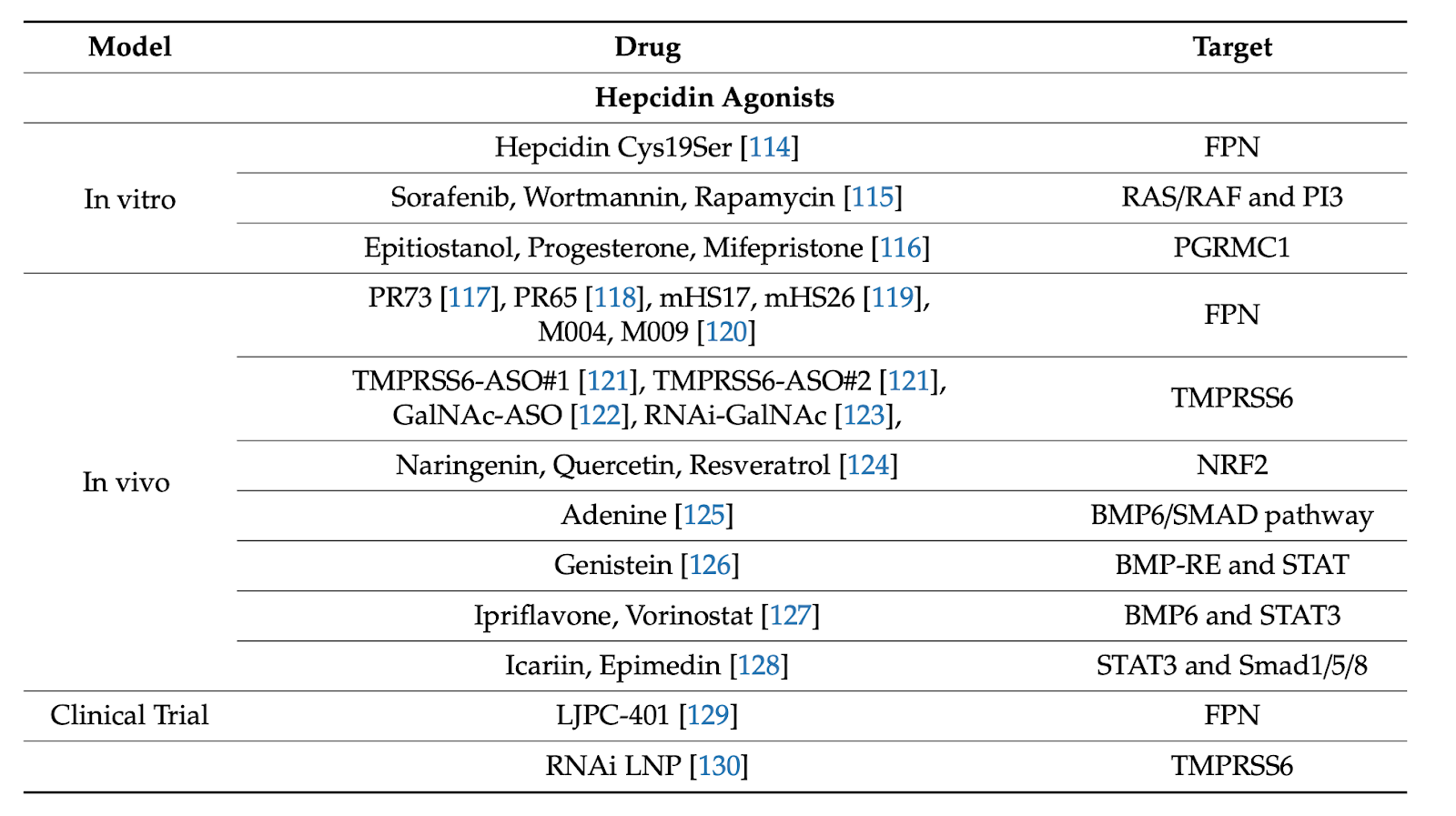
Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis, 2019
Hepcidin Therapeutics 2018
8.3. Small Molecule Hepcidin Inducers
Several small molecules were shown to activate hepcidin pathways. A high-throughput chemical
screen in zebrafish identified three steroid molecules i.e., progesterone, epitiostanol and mifepristone as potent hepcidin inducers. They operate via a pathway involving the progesterone receptor membrane component-1 (PRMC-1) [71]. A small-scale chemical screen in zebrafish embryos uncovered the hepcidin-inducing capacity of genistein, a phytoestrogen that is abundant in soybean. Genistein was shown to upregulate hepcidin in HepG2 hepatoma cells and zebrafish embryos by promoting STAT3 phosphorylation [72].
A subsequent high-throughput screen of small molecules in HepG2 cells identified ipriflavone
and vorinostat, which are synthetic drugs that inhibit bone resorption or histone deacetylase
activity and are clinically applied for the treatment of osteoporosis or cutaneous T cell lymphoma, respectively. They both stimulate expression of hepcidin and other BMP- and STAT3-dependent genes without affecting SMAD1/5/8 or STAT3 phosphorylation, and exhibit 10-fold higher potency than genistein [73]. Ipriflavone was further tested in vivo and was shown to alleviate dietary iron overload in wild type mice; however, it failed to reduce iron overload in Hbbth3/+ mice [74]. The hepcidin-stimulating effects of vorinostat in Huh7 hepatoma cells were also validated in another study [75], and are consistent with earlier data showing that the histone deacetylase inhibitor trichostatin A induces hepcidin by inhibiting the binding of C/EBPα and STAT3 in the HAMP promoter [76]. Nevertheless, vorinostat failed to induce hepcidin in mice [75]. The non-steroidal anti-inflammatory drug diclofenac likewise stimulated hepcidin expression in vitro, but not in vivo [75]. In a screen of natural compounds, icariin, a plant flavonol glycoside, was documented to stimulate hepcidin expression in HepG2 cells by inducing SMAD1/5/8 or STAT3 phosphorylation. These findings were validated in mice, while similar results were obtained with the icariin analogue epimedin C [77]. Several polyphenolic small molecules or phytoestrogens that are found in fruits and vegetables likewise induced hepcidin in HepG2 cells and in rats. These include resveratrol, quercetin, kaemferol, naringenin, epi-gallo-catechin-3-gallate, and operate by activating Nrf2 for binding to an
antioxidant response element (ARE) in the HAMP promoter [78].
A genetic siRNA screen revealed that Ras/Raf/MAPK and PI3K/Akt/mTOR signaling
suppress hepcidin; consequently, pharmacological inhibitors of these pathways, such as sorafenib, wortmannin, rapamycin and metformin were shown to induce hepcidin in hepatoma cells and primary hepatocytes [79]. In another setting, dietary supplementation of Hfe-/- mice with adenine was reported to induce hepcidin and attenuate iron overload by a mechanism requiring BMP/SMAD and cAMP/protein kinase A (PKA) signaling [80]. While some data obtained with small molecule hepcidin inducers are interesting, the potential for these drugs for clinical application in the context of hepcidin deficiency appears limited, due to lack of specificity.
Icariin regulates systemic iron metabolism by increasing hepatic hepcidin expression through Stat3 and Smad1/5/8 signaling, 2016
Systemic iron homeostasis is strictly controlled under normal conditions to ensure a balance between the absorption, utilization, storage and recycling of iron. The hepcidin-ferroportin (FPN) axis is of critical importance in the maintenance of iron homeostasis. Hepcidin deficiency gives rise to enhanced dietary iron absorption, as well as to increased iron release from macrophages, and this in turn results in iron accumulation in the plasma and organs, and is associated with a range of tissue pathologies. Low hepcidin levels have been demonstrated in most forms of hereditary hemochromatosis (HH), as well as in β-thalassemia. Therapies that increase hepcidin concentrations may potentially play a role in the treatment of these iron overload-related diseases. To date, natural compounds have not been extensively investigated for this purpose, to the best of our knowledge. Thus, in the present study, we screened natural compounds that have the potential to regulate hepcidin expression. By performing hepcidin promoter-luciferase assay, RT-qPCR and animal experiments, we demonstrated that icariin and berberine were potent stimulators of hepcidin transcription. Mechanistic experiments indicated that icariin and berberine increased hepcidin expression by activating the signal transducer and activator of transcription 3 (Stat3) and Smad1/5/8 signaling pathways. The induction of hepcidin was confirmed in mice following icariin administration, coupled with associated changes in serum and tissue iron concentrations. In support of these findings, the icariin analogues, epimedin A, B and C, also increased hepatic hepcidin expression. However, these changes were not observed in hepcidin-deficient [Hamp1-/- or Hamp1‑knockout (KO)] mice following icariin administration, thereby verifying hepatic hepcidin as the target of icariin. Although berberine exhibited a robust capacity to promote hepcidin expression in vitro, it failed to alter hepcidin expression in mice. Taken together, the findings of the present study suggest that icariin exhibits a robust capacity to increase hepatic hepcidin expression and to modulate systemic iron homeostasis. The present study therefore highlights the significance of using natural compounds to ameliorate iron disorders through the regulation of hepcidin expression.
Focus on phytosubstances – amazing properties of epimedium and icariin (link)
Icariin is the active flavinoid substance in the traditional Chinese medicinal herb Epimedium brevicornum Maxim. Icariin can be derived from several species of plants in the Epimedium family. These plants are known most popularly as Horny Goat Weed or Yin Yang Huo.[1]” Although known and marketed widely in the United States as an aphrodisiac(ref), icariin and its sister epimedium-derived flavinoids have been subect to extensive research, mainly in China and elsewhere in Asia, and have been shown to exhibit amazing health-producing properties. A series of in-vitro and animal model studies have shown that icariin can promote the differentiation and proliferation of cardiomyocytes and other stem cells in multiple organ systems, act as an antidepressant, be protective of neural cells, inhibit the breakdown of bone tissue, stimulate the development of new bone tissue, inhibit the actions of several toxic substances, attenuate unwanted microglial activation, stimulate angiogenesis, have a powerful effect in regulating the immune response, inhibit the inflammatory response in arthritis and other inflammatory disease conditions, and reduce or reverse bone loss due to injury or arthritis. Icariin administration extends the lifespan and healthspan of nematodes. It affects expression in numerous signaling pathways including MAPK, IGF-1, BMP, AMPK, NF-kappaB, MEK/ERK- and PI3K/Akt/eNOS, and, potentially could be the basis for new treatments addressing cancers, arthritis, osteoarthritis, asthma, acne, Alzheimer’s disease — and the list goes on. Finally, yes: older rats systematically administered icariin do exhibit accelerated sexual activity.
Along with quercetin, astragaloside IV, and ginsenoside, icariin may produce Cytochrome P450 drug interactions in a dose-dependent manner.
Mao H, Zhang L, Wang Y, et al. [Experimental studies of icariin on anticancer mechanism]. Zhong Yao Cai. Sep 2000;23(9):554-556.
Mao H, Zhang L, Wang Y, et al. [Influence of icariin on cell membrane of highly metastatic human lung tumor cell line]. Zhong Yao Cai. Jan 1999;22(1):35-36.
Guo Y, Zhang X, Meng J, et al. An anticancer agent icaritin induces sustained activation of the extracellular signal-regulated kinase (ERK) pathway and inhibits growth of breast cancer cells. Eur J Pharmacol. May 11 2011;658(2-3):114-122.
Horny Goat Weed Benefits (link)
In mice, icariin reduced physical fatigue and increased endurance during a weight-loaded swimming test [49].
In a clinical trial on 49 men, a non-prescription Japanese medicine with horny goat weed and 5 other herbal extracts improved several physical and psychological symptoms of aging [14].
icariin normalized levels of stress hormones – cortisol and CRF – in depressed rats under chronic mild stress. CRF
In one clinical trial of 90 postmenopausal women, an extract of horny goat weed increased estrogen levels and lowered blood cholesterol [27, 28].
A multi-herbal Chinese remedy with horny goat weed (Fufang) reduced bone loss and fractures in another trial on almost 200 postmenopausal women followed up for 5 years [23].
Horny goat weed may also enhance libido. In a study of 22 people with chronic kidney disease, it improved sex drive and quality of life [18].
In a clinical trial on 63 men with mild to moderate ED, a traditional Thai medicine with horny goat weed and 12 other herbs (Capra) improved erectile function and only caused mild adverse effects (the most common one being dizziness) [13].
icariin is a weak PDE5 inhibitor. Viagra (sildenafil), for example, is 80 times more effective at inhibiting PDE5 than icariin.
Interactions (link)
Cytochrome P450 substrates
Extended use of horny goat weed can increase the production of CYP3A4. Thus, it may reduce the effects of many drugs that are broken down by this group of enzymes [58].
Aromatase Inhibitors
Icariin increases the activity of aromatase, an enzyme involved in estrogen production. This can reduce the effect of aromatase inhibitors (e.g. anastrozole, exemestane, and letrozole) [10].
Blood-Pressure-Lowering Drugs
Horny goat weed may decrease blood pressure. Its combination with medications for high blood pressure might cause your blood pressure to drop too low [1].
Horny goat weed (Epimedium) is a herbal medicine that may comprise of mixtures of different related Epimedium species, which contain numerous and varied active constituents.
There is no published human information on drug interactions with horny goat weed. There is, however, the potential for significant drug interactions due to its inhibitory action on certain subgroups of the cytochrome P450 enzyme system, although the degree to which it does so is currently unknown. Drugs with which horny goat weed may interact via this mechanism include warfarin, amitriptyline, clarithromycin and erythromycin, ciclosporin and tacrolimus, and phenytoin. Furthermore, horny goat weed may enhance or prolong the effects of drugs that prolong the QT interval, and there is a potential for an additive interaction with phosphodiesterase‑5 inhibitors for erectile dysfunction.
Medications for high blood pressure (Antihypertensive drugs) interacts with HORNY GOAT WEED
Horny goat weed might lower blood pressure. Taking horny goat weed along with medications that lower blood pressure might cause blood pressure to go too low. Monitor your blood pressure closely.
Medications that slow blood clotting (Anticoagulant / Antiplatelet drugs) interacts with HORNY GOAT WEED
Horny goat weed might slow blood clotting. Taking horny goat weed along with medications that also slow blood clotting might increase the risk of bruising and bleeding.
Estrogens interacts with HORNY GOAT WEED
Horny goat weed might have some of the same effects as estrogen and might increase blood levels of estrogen in some people. Taking horny goat weed with estrogen might increase the effects and side effects of estrogen.
Medications changed by the liver (Cytochrome P450 1A2 (CYP1A2) substrates) interacts with HORNY GOAT WEED
Some medications are changed and broken down by the liver. Horny goat weed might change how quickly the liver breaks down these medications. This could change the effects and side effects of these medications.
Medications changed by the liver (Cytochrome P450 2B6 (CYP2B6) substrates) interacts with HORNY GOAT WEED
Some medications are changed and broken down by the liver. Horny goat weed might change how quickly the liver breaks down these medications. This could change the effects and side effects of these medications.
Interactions: There is conflicting evidence about a possible interaction with other PDE-5 inhibitors such as sildenafil (Viagra). In vitro evidence has suggested that epimedium potentiates PDE-5 inhibitors (Chiu et al. 2006), but a study of the effects of E. sagittatum extract on the pharmacokinetics of sildenafil demonstrated that the area under the concentration– time curve of sildenafil was significantly decreased in groups that received a high dose of epimedium extract, suggesting antagonistic effects (Hsueh et al. 2013). Co-administration with PDE-5 inhibitors should, therefore, be avoided until further information is available. Another in vitro study found that epimedium is a potent inhibitor of cytochrome P450 isoforms (including CYP1A2, CYP2C19, CYP2E1, CYP2C9, CYP3A4, CYP2D6) and NADPH-CYP reductase, indicating a possible potential for interaction with other drugs, although the clinical significance is not known (Liu et al. 2006).
Pharmacokinetics In vitro, freeze-dried aqueous extracts of Herba Epimedii have been found to have some inhibitory effect on the cytochrome P450 isoenzyme CYP1A2, an effect thought to be related to the quercetin content of the herb.1 Extracts of Herba Epimedii may also inhibit (in decreasing order of potency) CYP2C19, CYP2E1, CYP2C9, CYP3A4, and CYP2D6,1 but the clinical relevance of this has not been established. See flavonoids, page 186, for information on the pharmacokinetics of individual flavonoids present in epimedium.
Page 186 The potential for flavonoids to alter drug metabolism by cytochrome P450 isoenzymes, in particular, and also intestinal and hepatic drug transporters, such as P-glycoprotein, has been extensively investigated in vitro.5,7 There are also some animal studies, but few human clinical pharmacokinetic studies, and those that are available have generally used very high doses of the flavonoids. There is at present no reason to avoid flavonoids in the diet, or in the form of herbal medicines (most of which contain significant amounts of flavonoids naturally), and many positive reasons for including them. However, very high doses (such as the use of specific flavonoid supplements) could potentially alter the metabolism of other drugs that are substrates for CYP3A4 and/or P-glycoprotein, and increase the bioavailability of some drugs; for instance the statins, page 194, such as lovastatin and simvastatin; ciclosporin, page 190; benzodiazepines, page 189, such as midazolam; and digoxin, page 191

Comments